




DCA Dichloroacetate Breakthrough Anticancer Agent
Mary, an old patient in my office, called in last week to ask for advice about her husband, Jim. He had been quite healthy for many years, and recently noticed back pain. His primary care doctor ordered a CAT scan which showed a large lung mass (Red Arrow Above image) and destructive lesions in the spine. Biopsies confirmed the lung mass was indeed cancer, with metastatic spread to the thoracic vertebral bodies. Jim was referred to the local oncologist who started radiation and chemotherapy. Above Header Image CAT scan of lung cancer mass (Red Arrow) in left lung courtesy of wikimedia commons.
After a week of chemotherapy, Jim was miserable from the adverse effects of nausea, and vomiting, and loss of appetite. Jim felt so bad, he declined any further chemotherapy treatment. Mary asked if I had any suggestions.
Integrative Oncology Clinics in Canada
I had just returned from Seattle attending an Integrative Cancer Meeting organized by Paul Anderson ND director of a Seattle clinic, Advanced Medical Specialties. Dr Anderson is the author of “Out of the Box Cancer Therapies”(50), and a pioneer in the use in the orphan drug DCA (dichloroacetate), which shares synergy with a vitamin supplement called Poly-MVA.(46-52) Indeed, Dr Anderson declares:
“The combined use of DCA and Poly-MVA has been one of the truly big advances in integrative cancer therapies in the past 20 years” (121)
One of the keynote speakers at the meeting was Neil McKinney ND , director of Vital Victoria, an integrative cancer clinic in Victoria, British Columbia across the border from Seattle. Dr Mckinney’s program includes his own formulation called Mito-SAP, Nebulized DCA and R-ALA (available from York Downs Compounding Pharmacy in Toronto) , described in his hand outs. (25) During his talk , Dr Mckinney made the observation that patient outcomes improved dramatically with the addition of a botanical extract, Solomon’s Seal.(26-28)
Another Canadian integrative cancer doctor not attending the meeting , but prominently mentioned was Dr Akbar Khan, Director of the Medicor Clinic in Toronto. Dr Khan was a keynote speaker at the Oct 2018 ICIM Cancer Meeting in Minneapolis, and has published many case reports detailing his extensive experience with DCA as a cancer treatment.(66-71)
All three clinics are good options for people like Jim who have declined further conventional treatment because of the adverse effects. All three offer integrative oncology with DCA, (Dichloroacetate), given with supportive vitamins, thiamine (benfotiamine) and R-alpha lipoic acid, which can be taken individually or in a combination,Poly-MVA . These vitamins are necessary to avoid neuropathy induced by DCA. We will discuss this in more detail below. First, a little background on targeting cancer cell metabolism.
Metabolic Reprogramming of the Cancer Cell – Warburg Effect
Basic science of cancer cell biology reveals “Cancer is a metabolic disease“. In other words, cancer cells have a fundamental metabolic derangement, first observed in 1921 by Otto Warburg, and given his name: the “Warburg Effect”. Targeting the metabolic pathways of the cancer cell while leaving normal cells unharmed provides a safer and more effective cancer treatment, avoiding toxic effects of chemotherapy.
Warburg Effect – Aerobic Glycolysis
Normal cells have two quite different energy pathways, oxidative and non-oxidative. The oxidative pathway uses oxygen (aerobic in the mitochondria) and the non-oxidative pathway metabolizes glucose in the cytoplasm (anaerobic in the cytoplasm). Normal cells prefer the more efficient oxidative pathway (in the mitochondria). However the cancer cell has “switched off” the oxidative pathway in the mitochondria and instead prefers the non-oxidative (anaerobic) pathway in the cytoplasm, called Glycolysis. The cancer cells shift their energy production to the cytoplasm, a form of “fermentation”, also called “aerobic glycolysis” (the Warburg Effect).
“High lactate generation and low glucose oxidation, despite normal oxygen conditions, are commonly seen in cancer cells and tumors….Historically known as the Warburg effect.”(1)
“High lactate accumulation, despite adequate oxygen availability, is a metabolic pattern commonly associated with malignant transformation of the uncontrolled dividing cell. This metabolic phenotype, termed aerobic glycolysis and historically known as the Warburg effect, is characterized by high glycolytic rates and reduced mitochondrial oxidation.”(1)
“The unique metabolism of most solid tumors….all result in a switch in metabolism from mitochondria-based glucose oxidation (GO) to cytoplasm-based glycolysis even under normoxia, also known as the Warburg effect.”(4)
Reprogramming the Metabolic Switch with DCA
One might then ask the next logical question: What is the exact mechanism by which cancer cells “turn off the mitochondrial oxidative pathway, and how can we switch it back on? Many years after Warburg’s original discovery, molecular biology has clarified the metabolic reprogramming of the cancer cell. The key is an enzyme called PDC (pyruvate dehydrogenase complex), which sits at the control point for entry of pyruvate into the mitochondria. Cancer cells turn off PDC by upregulating PDK (pyruvate dehydrogenase kinase).(2)
“The mitochondrial pyruvate dehydrogenase complex (PDC) irreversibly decarboxylates pyruvate to acetyl coenzyme A, thereby linking glycolysis to the tricarboxylic acid cycle (oxidative in the mitochondria) and defining a critical step in cellular bioenergetics…. Inhibition of PDC activity by pyruvate dehydrogenase kinase (PDK)–mediated phosphorylation has been associated with the pathobiology of many disorders of metabolic integration, including cancer. Consequently, the PDC/PDK axis has long been a therapeutic target.“(2)
The “Achilles Heel” of the cancer cell is the PDC/PDK axis.(2) If we could somehow inhibit PDK (which then upregulates PDC), this would reverse the metabolic reprogramming of the cancer cell, and unlock the cancer cell from a state of apoptosis resistance. (note apoptosis= programmed cell death)(3,4) Dr Sutendra says in 2013:
“The unique metabolism of most solid tumors integrates many molecular and genetic proximal signals, which all result in a switch in metabolism from mitochondria-based glucose oxidation (GO) to cytoplasm-based glycolysis even under normoxia, also known as the Warburg effect....Therefore, by reversing this mitochondrial remodeling, it is possible to “unlock” these cells from a state of apoptosis resistance, selectively inducing cancer cell death”.(4)
 DCA Inhibits PDK – Inhibits Glycolysis
DCA Inhibits PDK – Inhibits Glycolysis
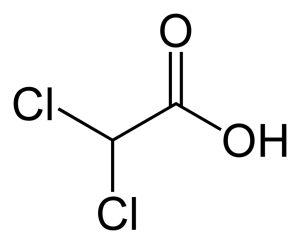
This is where DCA, dichloroacetate comes into play. DCA is an “orphan drug”, meaning it is old, off-patent drug originally used for lactic acidosis, and genetic mitochondrial disease.(8,9) Since DCA is off-patent and not profitable, drug companies are unlikely to fund large scale clinical trials. Even so, a number of clinical trials have been completed or are underway. Left image: Molecular Structure of DCA courtesy of wikimedia commons.
DCA May be Cancer Stem Cell Agent !! Excellent 2019 Review Article on DCA
Dr Tataranni suggests that DCA may have anticancer properties against cancer stem cells. See the 2019 excellent review article by Dr Tataranni in Med Cell Long (118) Dichloroacetate DCA and Cancer Overview Clinical Applications Tataranni Tiziana Oxidative Med Cel Longevity 2019
DCA Inhibits PDK
Remember, the cancer cell metabolism has been switched away from OXPHOS in the mitochondria to GLYCOLYSIS in the cytoplasm. The inhibition of PDK by DCA results in a “vastly diminished GLYCOLYSIS“ in the cancer cell. By inhibiting PDK, DCA switches the cancer cell metabolism back to oxidative phosphorylation (OXPhos) in the mitochondria. DCA is a “Glycolysis Inhibitor”, “forcing cancer cells to use mitochondria as the main ATP generator”.(3)
“One enzyme implicated in tumor metabolic remodeling and whose expression is regulated by oncogenic transcription factors is pyruvate dehydrogenase kinase 1 (PDK1), which is inhibited by dichloroacetate (DCA). PDK1 inhibition leads to pyruvate dehydrogenase (PDH) activation and forces cells to use mitochondria as the main ATP generator. As a result, glycolysis is vastly diminished.(3)
Translocation of Hexokinase II (HKII) from Cytoplasm to Mitochondria- HKII Binds to and Inhibits VDAC
Another prominent feature of of the metabolic reprogramming of the cancer cell involves the hexokinase enzyme, the first step in Glycolysis in the cytoplasm. Cancer cells have switched to an embryonic form of hexokinase called Hexokinase II, which then trans-locates from the cytoplasm to the outer mitochondrial membrane where it attaches to the VDAC (Voltage Dependent Anion Channel).(16-19) Hexokinase II is not present in normal cells, or only in small amounts. This is an important difference which can be targeted,
“In addition, the resultant concomitant up-regulation of glycolysis is associated with a translocation of hexokinase 2 from the cytoplasm to the mitochondrial membranes, where it has been shown to bind and further inhibit VDAC…“(4)
Cancer cells feature an embryonic form of Hexokinase II, normally not found in normal cells. Hexokinase normally resides in the cytoplasm, the first step in Glycolysis. Hexokinase II, however sits on the mitochondrial outer membrane the “pores” or “channels” called the VDAC, Voltage Dependent Anion Channells. Located here, the Hexokinase II gains access to energy in the form of ATP from the mitochondria, and blocks mitochondrial apoptosis, which “immortalizes” the cancer cell.
“Significantly, HK-2 is the major bound hexokinase isoform expressed in cancers that exhibit a “Warburg effect”. This includes most cancers that metastasize and kill their human host. By stationing itself on the outer mitochondrial membrane, HK-2 also helps immortalize cancer cells, escapes product inhibition and gains preferential access to newly synthesized ATP for phosphorylating glucose.(16)
Production of Lactic Acid and Acidity in the Micro-Environment
A byproduct of the Warburg Effect is the production of excess lactic acid which exits the cancer cells and creates an acidic microenvironment, involved in immune evasion. (16) “The resultant acidity likely wards off an immune response while preparing surrounding tissues for invasion.”(16) By reducing lactate acidity in the tumor microenvoironment, DCA restores anti-cancer host immune response. (141-143) Dr Ohashi says in 2013 IJC: “Dichloroacetate improves immune dysfunction caused by tumor‐secreted lactic acid and increases antitumor immunoreactivity.“
Schematic of the HK2/VDAC on The Mitochondrial Membrane
See the below diagram to see what this HK2-VDAC complex looks like:

Above Image: SChematic Diagram of the ATP Synthasome and HK/ VDAC Couple). (HK=Hexokinase II (Green) attached to VDAC (Blue) courtesy of Pederson and Ko (16) See Red Arrow in larger Diagram below for location on outer mitochondrial membrane:

Above Image: Red Arrow denotes VDAC/ HK2 complex with ATP Synthase.
Schematic courtesy of Pederson and Ko (16) Fig. 1 Metabolic channeling of glucose within a highly glycolytic tumor cell. Glucose brought across the plasma-membrane by glucose transporters is rapidly phosphorylated by HK-2 bound to VDAC located on the outer mitochondrial membrane (Red Arrow) . VDAC channels ATP generated by the ATP Synthasome complex (Red Arrow) (ATP synthase; adenine nucleotide translocator, ANT; inorganic phosphate carrier, PiC) on the inner mitochondrial membrane, facilitating direct access of ATP to VDAC-bound HK-2. To maintain the high rate of glycolytic metabolism in tumors, and their proliferation capacity, the product G-6-P rapidly distributes primarily across two key metabolic routes; (a) entry of G-6-P into the pentose–phosphate shunt for biosynthesis of nucleic-acid precursors, and (b) conversion of the G-6-P via the glycolytic pathway to pyruvic acid. Most of the pyruvic acid is reduced to lactic acid and transported out of the tumor cell via lactate transporters {B}. This promotes an unfavorable environment for the surrounding normal cells with concomitant regeneration of NAD+ within the cells to maintain glycolysis. Some pyruvate is directed to mitochondria across VDAC and via the “as-yet-uncharacterized” pyruvate transporter on the inner mitochondrial membrane {A}. This provides substrates for the tri-carboxylic acid (TCA) cycle for energy generation, as well as lipid and amino acid biosynthesis (not shown).Legend for Fig 1 (16)
Other Agents Which Target Mitochondrial Alterations of Cancer Cells
Below Image: Courtesy of Nguyen, Christopher, and Siyaram Pandey. “Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells.” Cancers 11.7 (2019). (22) This article lists all the drugs and compounds which target the Mitochondria: VDAC, HK2 and ETC (electron transport).

Itraconazole and Fenofibrate separates HK2 from VDAC
The attachment of Hexokinase II to the VDAC on the outer mitochondrial membrane creates a state of apoptosis resistance, and shunts ATP out from the mitochondria to the cytoplasm to support glycolysis.(16) It then follows logically that various repurposed drugs which separate Hexokinase II from the VDAC are effective “anti-cancer” drugs. Two such commonly available repurposed drugs are fenofibrate, a lipid lowering drug, and itraconazole, an antifungal drug. Both drugs are in common use. Dr Jan Chia-Ing writes that fenobrate is more potent than 3- bromopyruvate in disrupting HK2 from VDAC(73) :
“fenofibrate disrupted the interaction of hexokinase II and VDAC more potently than 3-BrPA (3-Bromo-Pyruvate) in both SAS and OECM1 (cancer) cells.” (SAS=human high-grade malignant oral cancer cell line and OECM1 = low-grade malignant oral cancer cell line)”(73)
See my previous article on Fenofibrate as Anti-cancer drug. as well as my previous article on Itraconazole as Anticancer drug.
Itraconazole Targets the VDAC on Outer Mitochondrial Membrane
Dr Sarah Head in 2015 used a flourescent probe to identify the VDAC as the molecular target of Itraconazole. DR Head found itraconazole is 1,000 times more effective than Metformin in activating AMPK.(23)
“In the present study we used a photoaffinity labeling approach using a biologically active itraconazole photoaffinity probe in live cells to identify the OMM (Outer Mitochondrial Membrane) channel VDAC1 as a molecular target of itraconazole.”(23)
“the concentrations of metformin required to activate AMPK in HUVEC are at least 1,000 times higher than those required of itraconazole (in the range of low millimoles) (55), suggesting that itraconazole might be significantly more effective than metformin at inhibiting angiogenesis in patients…..Itraconazole is pharmacologically distinct from other azole antifungal agents in that it is the only inhibitor in this class that has been shown to inhibit both the hedgehog signaling pathway[22][23] and angiogenesis.” (23)
In Blood 2016, Dr Juan J Gu studied Chemotherapy resistance NHL (non-hogkins lymphoma) cell lines in vitro and found itraconazole enhances the efficacy of chemotherapy agents.
“The disruption of HKII from mitochondria following itraconazole exposure may contribute to lower the mitochondrial membrane potential and enhance the chemotherapeutic efficacy. ” quote Dr Gu.
For more on itraconazole, see references (106-112)
 3 Bromo Pyruvate (3BP) Disassociates HK2 from VDAC (18-19)
3 Bromo Pyruvate (3BP) Disassociates HK2 from VDAC (18-19)
3 Bromo-Pyruvate is a pyruvate analog, a small molecule with anti-cancer activity studied by Drs Ko and Pederson.(18) Dr Chen in 2009 found that 3BP:
“directly triggered the dissociation of HKII from mitochondria, leading to a specific release of apoptosis-inducing factor (AIF) from the mitochondria to cytosol and eventual cell death.(19)
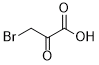
Disputes over patent rights may prevent this 3-BP drug from ever coming to practical use in the clinic. Upper Left Image : chemical structure of 3 Bromo Pyruvate courtesy of MedKoo Chemists
Lithium detaches HKII from mitochondria
Lithium is available at the vitamin shop as 5 mg lithium orotate capsules, as well as a prescription drug in the form of 300 lithium carbonate tablets used to treat bipolar disorder (note: checking serum lithium levels would be prodent when using lithium carbonate, as high levels can aproach toxicity). Dr Penso writes in 2003, that lithium detaches hexokinase from the mitochondria and inhibits melanoma (cancer) cells.(76)
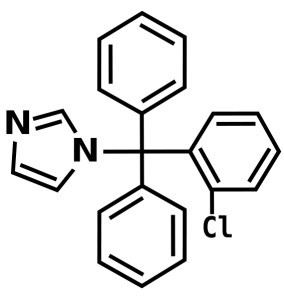 Clotrimazole
Clotrimazole
Left Image Chemical Structure of Clotrimiazole, courtesy of wikimedia commons
Another small molecule Clotrimazole, is an anti-fungal drug known to detach Hexokinase from the outer mitochondrial membrane. In 1998, Dr Penso writes the drug induces a dose-dependent detachment of hexokinase from the mitochondria of B16 melanoma cells. (74- 75)(22) in 2014, Dr Kadavakollu wrote an excellent review in Medicinal Chemistry of all the in-vitro, animal xenograft and clinical studies of Clotrimazole as anti-cancer drug.(74) In his article, Dr Kadavakollu summarizes efforts to enhance anti-cancer activity of Clotrimazole by forming a complex with rare earth metals such as palladium:
“The nitrogen atom present in the imidazole ring of clotrimazole facilitates coordination with transition metal ions such as Pt, as well as Ru, Pd, Cu, Co, Zn and Ni. Ravera et al. synthesized Pt(II) complexes containing bis(clotrimazole) ligands which were shown to effectively inhibit the growth of MCF-7, SKBR-3, HT-29, and B16/BL6 cell lines”(74)
With the above in mind, one might speculate synergy of Clotrimazole (and perhaps other azoles) with Poly-MVA which contains palladium in a liquid polymer with alpha lipoic acid and thiamine.
Methyl Jasmonate Detaches HKII from Mitochondria
Methyl Jasmonate is a plant “stress hormone” found in the Jasmine Plant. The oil is available and used widely in cosmetics, perfumes and aromatherapy. The methy jasmonate molecule has been extensively studied in vitro and xenograft animal models showing excellent profile with separation of HK2 from the Mitochondrial membrane, and induction of cancer cell death with virtually no toxic effects on normal cells. (77-93) Unfortunately, we still have yet to see any effort to develop a marketable drug or practical anti-cancer application of the natural substance.
Synergy of DCA with Electron Transport Chain Agents – Metformin ETC I
DCA Synergy with Defects in Electron Transport Chain
In 2010, Dr Luke Stockwin writes (12):
“Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC.(Electron Transport Chain”(12)
Metformin, a popular anti-diabetic drug used by millions, inhibits Complex I of the ETC (Electron Transport Chain).(94-95) Indeed, numerous studies show anti-cancer synergy of Metformin with DCA. The Metformin makes cancer cells more sensitive to DCA treatment. (21)(35-39)(113-117)
In 2015, Dr Seyfried writes:(21)
“Our data indicate that metformin enhancement of DCA cytotoxicity is dependent on complex I inhibition. Particularly, that complex I inhibition cooperates with DCA-induction of glucose oxidation to enhance cytotoxic oxidative stress in VM-M3 GBM (glioblastoma) cells.”(21)
In 2915 Oncotarget, Dr Ruggieri working with DCA and oral squamous carcinoma cells came to the same conclusion.(65)
“the therapeutic efficacy of DCA may depend on the specific metabolic profile adopted by the cancer cells with those exhibiting a deficient mitochondrial oxidative phosphorylation resulting more sensitive to the drug treatment.”(65)
DCA Synergy with Antibiotics Doxycycline and Clarithromycin ?
Another way to induce defects in the electron transport chain in mitochondria is with common antibiotics Doxycycline and Clarithromycin which inhibit bacterial (and therefore mitochondrial) ribosomal protein production, thus impairing the components of the Electron Transport chain. (123-130) An added bonus when targeting cancer cell mitochondria is the eradication of cancer stem cells which depend on OXPHOS. Dr Federica Sotgia writes in 2018 about repurposing antibiotics for targeting mitochondria in Cancer Stem Cells with the following commonly used antibiotics:(125)
a) Doxycycline;
b) Azithromycin;(Clarithromycin)
c) Pyrvinium (pamoate salt; not shown);
d) Atovaquone; and
e) Bedaquiline.
“Doxycycline and Azithromycin (a,b) are known to inhibit mitochondrial protein translation as an off-target side effect. They are used clinically as antibiotics to inhibit bacterial protein synthesis. Similarly, Pyrvinium pamoate and Atovaquone (c,d) are known to inhibit OXPHOS (related to mitochondrial complex II/III), as a side effect. Bedaquiline (e) was originally designed to inhibit the bacterial ATP-synthase, which is analogous to mitochondrial complex V. “(125)
Similar to the effect of Metformin, one might speculate that long term use of one or a combination of these antibiotics would interrupt mitochndrial function (inhibit OXPHOS), shunt metabolism towards glycolysis, and sensitize the cancer cells to DCA treatment.
See my previous articles on this:
Doxycycline and Vitamin C Anti Cancer Synergy
Clarithromycin Anticancer Antibiotic
Metformin Synergy with Venetoclax
Metformin was found to synergize with Venetoclax, a new oncology drug approved for use in Mantle Cell Lymphoma known to express high levels of the BCL-2 protein which inhibits apoptosis. (40) Venetoclax is commonly given in combination with Ibrutinib, a B-Cell Receptor Blocker, in Mantle Cell lymphoma and CLL.
Synergy Combination of DCA and Omaprazole PPI (10)
In 2012, Dr Ishiguro found that “Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors.” (10)
Anti-cancer effects of DCA synergizes with many other drugs and natural substances including:
Propranolol (32)
Celebrex (COX-2 Inhibitor)(34)
Salinomycin (Ivermectin?) (42)
Curcumin (44)
Phenylbutyrate(59)
Anti-Cancer Effects of DCA in Various Cancer Cells Lines:
Non-Hodgkins Lymphoma (29-30)
ColoRectal Cancer Cells(6)
Endometrial Cancer Cells (13)
Breast Cancer (14)
Pancreatic CA (31)
Ovarian Cancer(37)
Lung cancer (38)
Oral Squamous cell cancer(39)(65)
Multiple Myeloma (40)
Neuroblastoma (58)
Leukemia, Lymphoma, Multiple Myeloma(64)
T-Cell Lymphoma(Link)
Resensitizes Gastric CA to chemo (45)
Enhances Effects of chemotherapy (41)(118)
DCA Restores Host Immune Response by Inhibiting Lactic Acid Production
DCA reduction of lactic acidification in the tumor microenvironment helps to restore host anticancer immune response (141)
DCA has immunomodulatory activity, toward T1 function
Dr Badr writes in Oncotarget 2014:
“DCA has immunomodulatory activity, mainly via activation of the IL-12–IFN-gamma pathway and is able to modulate cytokines toward T helper 1 lymphocyte function.” (53)
Another 2913 study by Eleftheriadis DCA induced lymphocytes to differentiate into T-Regs which could result in decreased immunosurveillance (61) Whether this is a real issue or not is yet to be determined.
DCA inhibition of PDKII inhibits HIF1α in cancer cells (7)
This work suggests that mitochondria-targeting metabolic modulators (DCA) that increase pyruvate dehydrogenase activity, in addition to the recently described pro-apoptotic and anti-proliferative effects, suppress angiogenesis as well, normalizing the pseudo-hypoxic signals that lead to normoxic HIF1α activation in solid tumors.(7)
DCA has been studied in ALS (122) and chronic fatigue (43)
Adverse Side Effects of DCA
Neurotoxicity is a well known reversible adverse side effect of dichloroacetate, with a peripheral neuropathy with tingling and numbness in the extremities as the most common symptoms. (57) (96)(119) Neurotoxicity can be avoided by supplementing with Thiamine (Benfotiamine) , R-Alpha Lipoic Acid, reducing dosage of the DCA. Poly-MVA is also useful for reducing incidence of neuropathy. The neuropathy is reversible upon stopping the DCA. in 2016, a case report was published of a glioblastoma patient with fatal liver failure and bone marrow toxicity in combined use of DCA with Artesunate (56)
According to Dr Tataranni in 2019, DCA induced neuropathy is more common with the oral route, and can be avoided with IV DCA and the (inhaled) nebulized route.(118)
Conclusion: Overwhelming evidence has accumulated aver the years showing the metabolic approach with DCA a paradigm shift in cancer treatment. In spite of its promise, DCA is not routinely used, nor available on the oncology wards of US hospitals. When it becomes available is any one’s guess. Untill then, DCA treatment will be restricted to the integrative oncology practitioner, outside of conventional oncology.
Links to Articles with Similar Interest:
Anti-Cancer Activity of Natural Plant Pterostilbenes
Targeting Cancer Stem cells With Non-Toxic Therapies
Intravenous Vitamin C as Cancer Therapy
COX2 Inhibitor Celebrex as Anti-Cancer Drug
Metformin Repurposed Anticancer Drug
Doxycycline Vitamin C Anticancer Synergy
Jeffrey Dach MD
7450 Griffin Road Suote 180/190
Davie, Florida 33314
954-792-4663
references
1) McFate, Thomas, et al. “Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells.” Journal of Biological Chemistry 283.33 (2008): 22700-22708.
High lactate generation and low glucose oxidation, despite normal oxygen conditions, are commonly seen in cancer cells and tumors. Historically known as the Warburg effect, this altered metabolic phenotype has long been correlated with malignant progression and poor clinical outcome. However, the mechanistic relationship between altered glucose metabolism and malignancy remains poorly understood. Here we show that inhibition of pyruvate dehydrogenase complex (PDC) activity contributes to the Warburg metabolic and malignant phenotype in human head and neck squamous cell carcinoma. PDC inhibition occurs via enhanced expression of pyruvate dehydrogenase kinase-1 (PDK-1), which results in inhibitory phosphorylation of the pyruvate dehydrogenase α (PDHα) subunit. We also demonstrate that PDC inhibition in cancer cells is associated with normoxic stabilization of the malignancy-promoting transcription factor hypoxia-inducible factor-1α (HIF-1α) by glycolytic metabolites. Knockdown of PDK-1 via short hairpin RNA lowers PDHα phosphorylation, restores PDC activity, reverts the Warburg metabolic phenotype, decreases normoxic HIF-1α expression, lowers hypoxic cell survival, decreases invasiveness, and inhibits tumor growth. PDK-1 is an HIF-1-regulated gene, and these data suggest that the buildup of glycolytic metabolites, resulting from high PDK-1 expression, may in turn promote HIF-1 activation, thus sustaining a feed-forward loop for malignant progression. In addition to providing anabolic support for cancer cells, altered fuel metabolism thus supports a malignant phenotype. Correction of metabolic abnormalities offers unique opportunities for cancer treatment and may potentially synergize with other cancer therapies.
2) Stacpoole, Peter W. “Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer.” JNCI: Journal of the National Cancer Institute 109.11 (2017).
The mitochondrial pyruvate dehydrogenase complex (PDC) irreversibly decarboxylates pyruvate to acetyl coenzyme A, thereby linking glycolysis to the tricarboxylic acid cycle and defining a critical step in cellular bioenergetics. Inhibition of PDC activity by pyruvate dehydrogenase kinase (PDK)–mediated phosphorylation has been associated with the pathobiology of many disorders of metabolic integration, including cancer. Consequently, the PDC/PDK axis has long been a therapeutic target. The most common underlying mechanism accounting for PDC inhibition in these conditions is post-transcriptional upregulation of one or more PDK isoforms, leading to phosphorylation of the E1α subunit of PDC. Such perturbations of the PDC/PDK axis induce a “glycolytic shift,” whereby affected cells favor adenosine triphosphate production by glycolysis over mitochondrial oxidative phosphorylation and cellular proliferation over cellular quiescence. Dichloroacetate is the prototypic xenobiotic inhibitor of PDK, thereby maintaining PDC in its unphosphorylated, catalytically active form. However, recent interest in the therapeutic targeting of the PDC/PDK axis for the treatment of cancer has yielded a new generation of small molecule PDK inhibitors. Ongoing investigations of the central role of PDC in cellular energy metabolism and its regulation by pharmacological effectors of PDKs promise to open multiple exciting vistas into the biochemical understanding and treatment of cancer and other diseases.
3) Villalba, Martin, et al. “Chemical metabolic inhibitors for the treatment of blood-borne cancers.” Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) 14.2 (2014): 223-232.
One enzyme implicated in tumor metabolic remodeling and whose expression is regulated by oncogenic transcription factors is pyruvate dehydrogenase kinase 1 (PDK1), [3]), which is inhibited by dichloroacetate (DCA). PDK1 inhibition leads to pyruvate dehydrogenase (PDH) activation and forces cells to use mitochondria as the main ATP generator. As a result, glycolysis is vastly diminished.
4) Sutendra, Gopinath, and Evangelos D. Michelakis. “Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology.” Frontiers in oncology 3 (2013): 38.
Current drug development in oncology is non-selective as it typically focuses on pathways essential for the survival of all dividing cells. The unique metabolic profile of cancer, which is characterized by increased glycolysis and suppressed mitochondrial glucose oxidation (GO) provides cancer cells with a proliferative advantage, conducive with apoptosis resistance and even increased angiogenesis. Recent evidence suggests that targeting the cancer-specific metabolic and mitochondrial remodeling may offer selectivity in cancer treatment. Pyruvate dehydrogenase kinase (PDK) is a mitochondrial enzyme that is activated in a variety of cancers and results in the selective inhibition of pyruvate dehydrogenase, a complex of enzymes that converts cytosolic pyruvate to mitochondrial acetyl-CoA, the substrate for the Krebs’ cycle. Inhibition of PDK with either small interfering RNAs or the orphan drug dichloroacetate (DCA) shifts the metabolism of cancer cells from glycolysis to GO and reverses the suppression of mitochondria-dependent apoptosis. In addition, this therapeutic strategy increases the production of diffusible Krebs’ cycle intermediates and mitochondria-derived reactive oxygen species, activating p53 or inhibiting pro-proliferative and pro-angiogenic transcription factors like nuclear factor of activated T cells and hypoxia-inducible factor 1α. These effects result in decreased tumor growth and angiogenesis in a variety of cancers with high selectivity. In a small but mechanistic clinical trial in patients with glioblastoma, a highly aggressive and vascular form of brain cancer, DCA decreased tumor angiogenesis and tumor growth, suggesting that metabolic-targeting therapies can be translated directly to patients. More recently, the M2 isoform of pyruvate kinase (PKM2), which is highly expressed in cancer, is associated with suppressed mitochondrial function. Similar to DCA, activation of PKM2 in many cancers results in increased mitochondrial function and decreased tumor growth. Therefore, reversing the mitochondrial suppression with metabolic-modulating drugs, like PDK inhibitors or PKM2 activators holds promise in the rapidly expanding field of metabolic oncology.
The unique metabolism of most solid tumors integrates many molecular and genetic proximal signals, which all result in a switch in metabolism from mitochondria-based glucose oxidation (GO) to cytoplasm-based glycolysis even under normoxia, also known as the Warburg effect
it is now clear that this metabolic switch offers a survival advantage to cancer cells and a resistance to apoptosis, perhaps forming an essential pathway for cancer, but not normal cells. Therefore, by reversing this mitochondrial remodeling, it is possible to “unlock” these cells from a state of apoptosis resistance, selectively inducing cancer cell death. A critical mitochondrial enzyme and a gatekeeper of GO is pyruvate dehydrogenase (PDH), which exists in a complex with its inhibitor, PDH kinase (PDK). There is now evidence that several oncogenes or transcription factors critical for cancer progression, like loss of p53 (Contractor and Harris, 2012) or activation of hypoxia-inducible factor 1α (HIF1α; Kim et al., 2006), can induce PDK expression and thus inhibit PDH and GO.
In summary, there are multiple ways in which the metabolism of cancer cells (increased glycolysis and suppressed GO) is associated with mitochondrial remodeling (∆ψm hyperpolarization, decreased mROS production) that leads to resistance to apoptosis. A potentially simple way this metabolic profile and the suppression of apoptosis and redox signaling can be reversed is by the activation of the PDH, the gate-keeping regulator of GO. One particularly simple way is by the use of dichloroacetate (DCA).
In addition, the resultant concomitant up-regulation of glycolysis is associated with a translocation of hexokinase 2 from the cytoplasm to the mitochondrial membranes, where it has been shown to bind and further inhibit VDAC
administered DCA orally at a dose of 12.5 mg/kg twice a day in five patients with primary GBM
DCA “unlocks” cancer cells from a state of apoptosis resistance.
5) Rodrigues, Ana Sofia, et al. “Dichloroacetate, the pyruvate dehydrogenase complex and the modulation of mESC pluripotency.” PloS one 10.7 (2015): e0131663.
DCA is known for inhibiting all PDHK isoforms and it as been used in clinical trials for several types of tumors (lung, endometrial and breast cancer[12]) and other clinical conditions such as type II diabetes [15], congestive heart failure and congenital mitochondrial diseases [12] due to side effect of lowering lactate levels by activating the PDH complex.
DCA has been shown to affect cell metabolism by inhibiting PDHK, with a concomitant cellular shift from glycolysis to oxidative phosphorylation
DCA and ColoRectal Cancer Cells
6) Madhok, B. M., et al. “Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells.” British journal of cancer 102.12 (2010): 1746.
Cancer cells are highly dependent on glycolysis. Our aim was to determine if switching metabolism from glycolysis towards mitochondrial respiration would reduce growth preferentially in colorectal cancer cells over normal cells, and to examine the underlying mechanisms.
Representative colorectal cancer and non-cancerous cell lines were treated with dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase.
Dichloroacetate (20 mM) did not reduce growth of non-cancerous cells but caused significant decrease in cancer cell proliferation (P=0.009), which was associated with apoptosis and G2 phase cell-cycle arrest. The largest apoptotic effect was evident in metastatic LoVo cells, in which DCA induced up to a ten-fold increase in apoptotic cell counts after 48 h. The most striking G2 arrest was evident in well-differentiated HT29 cells, in which DCA caused an eight-fold increase in cells in G2 phase after 48 h. Dichloroacetate reduced lactate levels in growth media and induced dephosphorylation of E1α subunit of pyruvate dehydrogenase complex in all cell lines, but the intrinsic mitochondrial membrane potential was reduced in only cancer cells (P=0.04).
Pyruvate dehydrogenase kinase inhibition attenuates glycolysis and facilitates mitochondrial oxidative phosphorylation, leading to reduced growth of colorectal cancer cells but not of non-cancerous cells.
Most solid tumors are characterized by a metabolic shift from glucose oxidation to glycolysis, in part due to actively suppressed mitochondrial function, a state that favors resistance to apoptosis. Suppressed mitochondrial function may also contribute to the activation of hypoxia-inducible factor 1α (HIF1α) and angiogenesis. We have previously shown that the inhibitor of pyruvate dehydrogenase kinase (PDK) dichloroacetate (DCA) activates glucose oxidation and induces apoptosis in cancer cells in vitro and in vivo. We hypothesized that DCA will also reverse the ‘pseudohypoxic’ mitochondrial signals that lead to HIF1α activation in cancer, even in the absence of hypoxia and inhibit cancer angiogenesis. We show that inhibition of PDKII inhibits HIF1α in cancer cells using several techniques, including HIF1α luciferase reporter assays. Using pharmacologic and molecular approaches that suppress the prolyl-hydroxylase (PHD)-mediated inhibition of HIF1α, we show that DCA inhibits HIF1α by both a PHD-dependent mechanism (that involves a DCA-induced increase in the production of mitochondria-derived α-ketoglutarate) and a PHD-independent mechanism, involving activation of p53 via mitochondrial-derived H(2)O(2), as well as activation of GSK3β. Effective inhibition of HIF1α is shown by a decrease in the expression of several HIF1α regulated gene products as well as inhibition of angiogenesis in vitro in matrigel assays. More importantly, in rat xenotransplant models of non-small cell lung cancer and breast cancer, we show effective inhibition of angiogenesis and tumor perfusion in vivo, assessed by contrast-enhanced ultrasonography, nuclear imaging techniques and histology. This work suggests that mitochondria-targeting metabolic modulators that increase pyruvate dehydrogenase activity, in addition to the recently described pro-apoptotic and anti-proliferative effects, suppress angiogenesis as well, normalizing the pseudo-hypoxic signals that lead to normoxic HIF1α activation in solid tumors.
8) free pdf
Stacpoole, Peter W., et al. “Treatment of congenital lactic acidosis with dichloroacetate.” Archives of disease in childhood 77.6 (1997): 535-541.
9) Stacpoole, Peter W., et al. “Role of dichloroacetate in the treatment of genetic mitochondrial diseases.” Advanced drug delivery reviews 60.13-14 (2008): 1478-1487.
Synergy Combination of DCA and Omaprazole PPI
10) Ishiguro, Tatsuaki, et al. “Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors.” Oncology letters 3.3 (2012): 726-728.
It has been reported that treating cancer cells with dichloroacetate (DCA), an approved treatment for congenital lactic acidosis, reverses the Warburg effect and inhibits tumor growth). Furthermore, omeprazole (OMP) is a well-known agent that enhances the effects of anticancer drugs. The aim of this study was to find clinically-used drugs that enhance the effects of DCA. The combination of DCA and OMP exhibited a more potent antitumor activity than DCA alone in HT1080 fibrosarcoma cells and RKO colon cancer cells, while the drugs did not affect the proliferation of WI-38 human fibroblasts. The inhibitory effect of DCA combined with OMP was reversed with vitamin E and Z-VAD-FMK; therefore conventional caspase-dependent cell growth inhibition through superoxide production was suggested as the mechanism for inhibition. The combination of these drugs also had an effect on HT1080 fibrosarcoma cells inoculated into mice. Since OMP and DCA may be administered orally and have been used clinically for several years without major side effects, we believe that this combination therapy could be readily translated to treat malignant tumors.
11) Papandreou, Ioanna, Tereza Goliasova, and Nicholas C. Denko. “Anticancer drugs that target metabolism: is dichloroacetate the new paradigm?.” International journal of cancer 128.5 (2011): 1001-1008.Anticancer drugs target metabolism dichloroacetate new paradigm Papandreou 2011 Int J Cancer
12) Stockwin, Luke H., et al. “Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC.” International journal of cancer 127.11 (2010): 2510-2519.Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC Stockwin Luke Int j cancer 2010
The “Warburg effect,” also termed aerobic glycolysis, describes the increased reliance of cancer cells on glycolysis for ATP production, even in the presence of oxygen. Consequently, there is continued interest in inhibitors of glycolysis as cancer therapeutics. One example is dichloroacetate (DCA), a pyruvate mimetic that stimulates oxidative phosphorylation through inhibition of pyruvate dehydrogenase kinase. In this study, the mechanistic basis for DCA anti-cancer activity was re-evaluated in vitro using biochemical, cellular and proteomic approaches. Results demonstrated that DCA is relatively inactive (IC(50) ≥ 17 mM, 48 hr), induces apoptosis only at high concentrations (≥ 25 mM, 48 hr) and is not cancer cell selective. Subsequent 2D-PAGE proteomic analysis confirmed DCA-induced growth suppression without apoptosis induction. Furthermore, DCA depolarizes mitochondria and promotes reactive oxygen species (ROS) generation in all cell types. However, DCA was found to have selective activity against rho(0) cells [mitochondrial DNA (mtDNA) deficient] and to synergize with 2-deoxyglucose in complex IV deficient HCT116 p53(-/-) cells. DCA also synergized in vitro with cisplatin and topotecan, two antineoplastic agents known to damage mitochondrial DNA. These data suggest that in cells “hardwired” to selectively utilize glycolysis for ATP generation (e.g., through mtDNA mutations), the ability of DCA to force oxidative phosphorylation confers selective toxicity. In conclusion, although we provide a mechanism distinct from that reported previously, the ability of DCA to target cell lines with defects in the electron transport chain and to synergize with existing chemotherapeutics supports further preclinical development.
13) Wong, Jason YY, et al. “Dichloroacetate induces apoptosis in endometrial cancer cells.” Gynecologic oncology 109.3 (2008): 394-402.
14) Sun, Ramon C., et al. “Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo.” Breast cancer research and treatment 120.1 (2010): 253-260.Reversal of glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer Sun Ramon Breast cancer res 2010
15) Michelakis, E. D., L. Webster, and J. R. Mackey. “Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer.” British journal of cancer 99.7 (2008): 989. University of Alberta, Edmonton, Canada
The unique metabolism of most solid tumours (aerobic glycolysis, i.e., Warburg effect) is not only the basis of diagnosing cancer with metabolic imaging but might also be associated with the resistance to apoptosis that characterises cancer. The glycolytic phenotype in cancer appears to be the common denominator of diverse molecular abnormalities in cancer and may be associated with a (potentially reversible) suppression of mitochondrial function. The generic drug dichloroacetate is an orally available small molecule that, by inhibiting the pyruvate dehydrogenase kinase, increases the flux of pyruvate into the mitochondria, promoting glucose oxidation over glycolysis. This reverses the suppressed mitochondrial apoptosis in cancer and results in suppression of tumour growth in vitro and in vivo.
Hexokinase II pivotal role-Good diagram !!!!!
16) Mathupala, Saroj P., Young H. Ko, and Peter L. Pedersen. “Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy.” Seminars in cancer biology. Vol. 19. No. 1. Academic Press, 2009.
The most common metabolic hallmark of malignant tumors, i.e., the “Warburg effect” is their propensity to metabolize glucose to lactic acid at a high rate even in the presence of oxygen. The pivotal player in this frequent cancer phenotype is mitochondrial-bound hexokinase
17) Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem 1981;256(16):8699−704].
Now, in clinics worldwide this prominent phenotype forms the basis of one of the most common detection systems for cancer, i.e., positron emission tomography (PET). Significantly, HK-2 is the major bound hexokinase isoform expressed in cancers that exhibit a “Warburg effect”. This includes most cancers that metastasize and kill their human host. By stationing itself on the outer mitochondrial membrane, HK-2 also helps immortalize cancer cells, escapes product inhibition and gains preferential access to newly synthesized ATP for phosphorylating glucose. The latter event traps this essential nutrient inside the tumor cells as glucose-6-P, some of which is funneled off to serve as carbon precursors to help promote the production of new cancer cells while much is converted to lactic acid that exits the cells. The resultant acidity likely wards off an immune response while preparing surrounding tissues for invasion. With the re-emergence and acceptance of both the “Warburg effect” as a prominent phenotype of most clinical cancers, and “metabolic targeting” as a rational therapeutic strategy, a number of laboratories are focusing on metabolite entry or exit steps. One remarkable success story
18) Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, et al. Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun 2004;324(1):269−75]
is the use of the small molecule 3-bromopyruvate (3-BP) that selectively enters and destroys the cells of large tumors in animals by targeting both HK-2 and the mitochondrial ATP synthasome. This leads to very rapid ATP depletion and tumor destruction without harm to the animals. This review focuses on the multiple roles played by HK-2 in cancer and its potential as a metabolic target for complete cancer destruction.
19) Chen, Zhao, et al. “Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate.” Biochimica et Biophysica Acta (BBA)-Bioenergetics 1787.5 (2009): 553-560.
It has long been observed that cancer cells rely more on glycolysis to generate ATP and actively use certain glycolytic metabolic intermediates for biosynthesis. Hexokinase II (HKII) is a key glycolytic enzyme that plays a role in the regulation of the mitochondria-initiated apoptotic cell death. As a potent inhibitor of hexokinase, 3-bromopyruvate (3-BrPA) is known to inhibit cancer cell energy metabolism and trigger cell death, supposedly through depletion of cellular ATP. The current study showed that 3-BrPA caused a covalent modification of HKII protein and directly triggered its dissociation from mitochondria, leading to a specific release of apoptosis-inducing factor (AIF) from the mitochondria to cytosol and eventual cell death. Co-immunoprecipitation revealed a physical interaction between HKII and AIF. Using a competitive peptide of HKII, we showed that the dissociation of hexokinase II from mitochondria alone could cause apoptotic cell death, especially in the mitochondria-deficient rho(0) cells that highly express HKII. Interestingly, the dissociation of HKII itself did not directly affect the mitochondrial membrane potential, ROS generation, and oxidative phosphorylation. Our study suggests that the physical association between HKII and AIF is important for the normal localization of AIF in the mitochondria, and disruption of this protein complex by 3-BrPA leads to their release from the mitochondria and eventual cell death.
20) Bhat, Tariq A., et al. “Restoration of mitochondria function as a target for cancer therapy.” Drug discovery today 20.5 (2015): 635-643.
21) Seyfried, Thomas N. “Cancer as a mitochondrial metabolic disease.” Frontiers in cell and developmental biology 3 (2015): 43.
Cancer is widely considered a genetic disease involving nuclear mutations in oncogenes and tumor suppressor genes. This view persists despite the numerous inconsistencies associated with the somatic mutation theory. In contrast to the somatic mutation theory, emerging evidence suggests that cancer is a mitochondrial metabolic disease, according to the original theory of Otto Warburg. The findings are reviewed from nuclear cytoplasm transfer experiments that relate to the origin of cancer. The evidence from these experiments is difficult to reconcile with the somatic mutation theory, but is consistent with the notion that cancer is primarily a mitochondrial metabolic disease.
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
2019 Targeting HKII -VDAC with Fenofibrate, Clotrimazole, DCA, Oroxylyn (Baical)
Niuce Recent Summary Review
22) Nguyen, Christopher, and Siyaram Pandey. “Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells.” Cancers 11.7 (2019).
Cancer cells rely heavily on aerobic glycolysis and, as a result, over-express hexokinases (HK) that catalyze the first step of glucose metabolism [131,132]. Of the four HK isoforms, hexokinase II (HKII) plays a critical role in cancer cell survival and proliferation. At the outer mitochondrial membrane, HKII binds to voltage-dependent anion channel 1 (VDAC1) and facilitates its interaction with adenine nucleotide translocase on the inner mitochondrial membrane [133,134]. This interaction is able to couple aerobic glycolysis with OxPhos and create a working relationship between the two metabolic processes, allowing HKII to exchange ADP for ATP from the mitochondria and increase the rate of glycolysis [72,134]. Targeting HKII could lead to uncoupling and stunting of aerobic glycolysis, leading to cancer cell death.
An antifungal drug, itraconazole, was shown to target VDAC1 and inhibit mTOR activity and cell proliferation through modulation of mitochondrial metabolism, leading to apoptosis [88]
the VDAC1–HKII complex can be targeted to trigger apoptosis in cancer cells overexpressing HKII. In a recent study, fenofibrate interrupted the binding of HKII to VDAC1 and reprogrammed the metabolic pathway in oral squamous cell carcinoma
Itraconazole Targets VDAC – Modulates mTOR
23) Head, Sarah A., et al. “Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells.” Proceedings of the National Academy of Sciences 112.52 (2015): E7276-E7285.
“Itraconazole is pharmacologically distinct from other azole antifungal agents in that it is the only inhibitor in this class that has been shown to inhibit both the hedgehog signaling pathway[22][23] and angiogenesis.[24][25]”
————— ———-
Survey of all the molecules, peptides, and microRNAs that exploit VDAC
24) Magrì, Andrea, Simona Reina, and Vito De Pinto. “VDAC1 as pharmacological target in cancer and neurodegeneration: focus on its role in apoptosis.” Frontiers in chemistry 6 (2018): 108.
Cancer and neurodegeneration are different classes of diseases that share the involvement of mitochondria in their pathogenesis. Whereas the high glycolytic rate (the so-called Warburg metabolism) and the suppression of apoptosis are key elements for the establishment and maintenance of cancer cells, mitochondrial dysfunction and increased cell death mark neurodegeneration. As a main actor in the regulation of cell metabolism and apoptosis, VDAC may represent the common point between these two broad families of pathologies. Located in the outer mitochondrial membrane, VDAC forms channels that control the flux of ions and metabolites across the mitochondrion thus mediating the organelle’s cross-talk with the rest of the cell. Furthermore, the interaction with both pro-apoptotic and anti-apoptotic factors makes VDAC a gatekeeper for mitochondria-mediated cell death and survival signaling pathways. Unfortunately, the lack of an evident druggability of this protein, since it has no defined binding or active sites, makes the quest for VDAC interacting molecules a difficult tale. Pharmacologically active molecules of different classes have been proposed to hit cancer and neurodegeneration. In this work, we provide an exhaustive and detailed survey of all the molecules, peptides, and microRNAs that exploit VDAC in the treatment of the two examined classes of pathologies. The mechanism of action and the potential or effectiveness of each compound are discussed.
=======================================================
25) Dr Neil McKinney Handouts
B) ALA – R vs racemic Neil McKinney2
C) Metabolic Approach to CancerNeil McKinney 3
D) Nebulizing ALA & DCA McKinney 4
E) A NUTRITION BASED STRATEGY FOR CONTROLLING CANCER CELL ENERGETICS McKinney 5
Solomon’s Seal – Dr Neil McKinney
26) Wang, Shu-ya, et al. “Polygonatum cyrtonema lectin, a potential antineoplastic drug targeting programmed cell death pathways.” Biochemical and biophysical research communications 406.4 (2011): 497-500. pdf link: Polygonatum cyrtonema lectin potential antineoplastic drug targeting programmed cell death pathways Solomon Seal 2011
Polygonatum cyrtonema lectin (PCL), a mannose/sialic acid-binding plant lectin, has recently drawn a rising attention for cancer biologists because PCL bears remarkable anti-tumor activities and thus inducing programmed cell death (PCD) including apoptosis and autophagy in cancer cells. In this review, we focus on exploring the precise molecular mechanisms by which PCL induces cancer cell apoptotic death such as the caspase-dependent pathway, mitochondria-mediated ROS-p38-p53 pathway, Ras-Raf and PI3K-Akt pathways. In addition, we further elucidate that PCL induces cancer cell autophagic death via activating mitochondrial ROS-p38-p53 pathway, as well as via blocking Ras-Raf and PI3K-Akt pathways, suggesting an intricate relationship between autophagic and apoptotic death in PCL-induced cancer cells. In conclusion, these findings may provide a new perspective of Polygonatum cyrtonema lectin (PCL) as a potential anti-tumor drug targeting PCD pathways for future cancer therapeutics.
Solomons seal and MC-7 Breast cancer Cells
27) Ouyang, Liang, et al. “Polygonatum odoratum lectin induces apoptosis and autophagy via targeting EGFR-mediated Ras-Raf-MEK-ERK pathway in human MCF-7 breast cancer cells.” Phytomedicine 21.12 (2014): 1658-1665.
Polygonatum odoratum lectin (POL), a mannose-binding GNA-related lectin, has been reported to display remarkable anti-proliferative and apoptosis-inducing activities toward a variety of cancer cells; however, the precise molecular mechanisms by which POL induces cancer cell death are still elusive. In the current study, we found that POL could induce both apoptosis and autophagy in human MCF-7 breast cancer cells. Subsequently, we found that POL induced MCF-7 cell apoptosis via the mitochondrial pathway. Additionally, we also found that POL induces MCF-7 cell apoptosis via EGFR-mediated Ras-Raf-MEK-ERK pathway, suggesting that POL may be a potential EGFR inhibitor. Finally, we used proteomics analyses for exploring more possible POL-induced pathways with EGFR, Ras, Raf, MEK and ERK, some of which were consistent with our in silico network prediction. Taken together, these results demonstrate that POL induces MCF-7 cell apoptosis and autophagy via targeting EGFR-mediated Ras-Raf-MEK-ERK signaling pathway, which would provide a new clue for exploiting POL as a potential anti-neoplastic drug for future cancer therapy.
Solomons Seal and Lung Cancer – Localized on Mitochondia
28) Liu, Tao, et al. “Role of reactive oxygen species-mediated MAPK and NF-κB activation in polygonatum cyrtonema lectin-induced apoptosis and autophagy in human lung adenocarcinoma A549 cells.” The Journal of Biochemistry 160.6 (2016): 315-324.
In a previous study, Chang et al. reported that legume lectin Concanavalin A (ConA) bound to cell membrane glycoproteins and was internalized onto the mitochondria in hepatoma ML-14a cells (19). However, it is not clear whether PCL can also get into the A549 cells. Thus, we used the FITC-PCL to track its subcellular distribution (Fig. 6D). PCL was found to localize on the mitochondria of the A549 cells as shown by the merger of fluorescence of PCL and fluorescence of Mito Tracker Red. Only a small percent of the labelled PCL was found in the lysosome and the endoplasmic reticulum, suggesting PCL was mainly localized on the mitochondria. These results suggest that PCL could bind to A549 cells in a mannose-specific manner and then localized on the mitochondria
In this study, we report here that PCL induces apoptosis and autophagy via ROS mediated MAPK and NF-κB signalling pathways in A549 cells. Importantly, PCL was found cytotoxic to the A549 cells and exhibited low cytotocity toward HELF cells, suggesting PCL is endowed with a number of favourable and interesting properties that make it as the primary candidate drug to be considered for further (pre)clinical investigations as potential anti-tumour agents.
—————— ————————————
DCA NHL Reversal
29) Flavin, Dana F. “Non-Hodgkin’s lymphoma reversal with dichloroacetate.” Journal of oncology 2010 (2010).
In June 2007, a 48-year-old male patient, diagnosed with Stage 4 Non-Hodgkin’s Follicular Lymphoma (NHL), was treated for 3 months with conventional chemotherapy resulting in a complete remission. Almost one year later tumors returned in the nasopharynx and neck lymph glands. Refusing all suggested chemotherapies, the patient began self-administering dichloroacetate (DCA) 900 mg daily with a PET scan showing complete remission four months later. Since his last PET scan, May, 2009, he remains tumor-free from continuous DCA usage.
First and foremost DCA inhibits pyruvate dehydrogenase kinase (PDK). PDK blocks pyruvate dehydrogenase (PDH) through its phosphorylation activity.
DCA has been shown to block this phosphorylation by PDK at the mitochondrial membrane level and decrease glycolysis in favor of glucose oxidation. This return to a normal metabolism of glucose allows for major changes including a decrease in Ca++ intracellularly, and stabilization of the mitochondria allowing a reactivation of caspases in cancer cells leading to apoptosis [19]
The effects of DCA, caused by reactivation of mitochondrial respiration, are not without complications although it inexplicably seems to be predominantly limited to cancer cells while most normal cells remain unaffected [24]. A reversible, minimal nerve damage can be considerably reduced by a daily thiamine intake of several hundred milligrams for humans [23] and animals [15]. The thiamine amount varies from 50 mg/day to 100 mg/day depending on whether it is administered orally or injected intramuscularly [23].
DCA Complete Response in NHL AFter Chemo Relapse
30) Strum, Stephen B., et al. “Case Report: Sodium dichloroacetate (DCA) inhibition of the “Warburg Effect” in a human cancer patient: complete response in non-Hodgkin’s lymphoma after disease progression with rituximab-CHOP.” (2012).dichloroacetate DCA) inhibition of Warburg Effect complete response in non-Hodgkin’s lymphoma Strum 2012
DCA and Pancreatic Cancer
31) Tataranni, Tiziana, et al. “Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines.” Cells 8.5 (2019): 478.
Targeting metabolism represents a possible successful approach to treat cancer. Dichloroacetate (DCA) is a drug known to divert metabolism from anaerobic glycolysis to mitochondrial oxidative phosphorylation by stimulation of PDH.
Targeting metabolism represents a possible successful approach to treat cancer. Dichloroacetate (DCA) is a drug known to divert metabolism from anaerobic glycolysis to mitochondrial oxidative phosphorylation by stimulation of PDH. In this study, we investigated the response of two pancreatic cancer cell lines to DCA, in two-dimensional and three-dimension cell cultures, as well as in a mouse model. PANC-1 and BXPC-3 treated with DCA showed a marked decrease in cell proliferation and migration which did not correlate with enhanced apoptosis indicating a cytostatic rather than a cytotoxic effect. Despite PDH activation, DCA treatment resulted in reduced mitochondrial oxygen consumption without affecting glycolysis. Moreover, DCA caused enhancement of ROS production, mtDNA, and of the mitophagy-marker LC3B-II in both cell lines but reduced mitochondrial fusion markers only in BXPC-3. Notably, DCA downregulated the expression of the cancer stem cells markers CD24/CD44/EPCAM only in PANC-1 but inhibited spheroid formation/viability in both cell lines. In a xenograft pancreatic cancer mouse-model DCA treatment resulted in retarding cancer progression. Collectively, our results clearly indicate that the efficacy of DCA in inhibiting cancer growth mechanistically depends on the cell phenotype and on multiple off-target pathways. In this context, the novelty that DCA might affect the cancer stem cell compartment is therapeutically relevant.
————————- –
2018 DCA combined with propranolol
32) Lucido, Christopher T., W. Keith Miskimins, and Paola D. Vermeer. “Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC.” Cancers 10.12 (2018).
Tumor cell metabolism differs from that of normal cells, conferring tumors with metabolic advantages but affording opportunities for therapeutic intervention. Accordingly, metabolism-targeting therapies have shown promise. However, drugs targeting singular metabolic pathways display limited efficacy, in part due to the tumor’s ability to compensate by using other metabolic pathways to meet energy and growth demands. Thus, it is critical to identify novel combinations of metabolism-targeting drugs to improve therapeutic efficacy in the face of compensatory cellular response mechanisms. Our lab has previously identified that the anti-cancer activity of propranolol, a non-selective beta-blocker, is associated with inhibition of mitochondrial metabolism in head and neck squamous cell carcinoma (HNSCC). In response to propranolol, however, HNSCC exhibits heightened glycolytic activity, which may limit the effectiveness of propranolol as a single agent. Thus, we hypothesized that propranolol’s metabolic effects promote a state of enhanced glucose dependence, and that propranolol together with glycolytic inhibition would provide a highly effective therapeutic combination in HNSCC. Here, we show that glucose deprivation synergizes with propranolol for anti-cancer activity, and that the rational combination of propranolol and dichloroacetate (DCA), a clinically available glycolytic inhibitor, dramatically attenuates tumor cell metabolism and mTOR signaling, inhibits proliferation and colony formation, and induces apoptosis. This therapeutic combination displays efficacy in both human papillomavirus-positive (HPV(+)) and HPV(−) HNSCC cell lines, as well as a recurrent/metastatic model, while leaving normal tonsil epithelial cells relatively unaffected. Importantly, the combination significantly delays tumor growth in vivo with no evidence of toxicity. Additionally, the combination of propranolol and DCA enhances the effects of chemoradiation and sensitizes resistant cells to cisplatin and radiation. This novel therapeutic combination represents a promising treatment strategy which may overcome some of the limitations of targeting individual metabolic pathways in cancer.
33) Pantziarka, Pan, et al. “Repurposing Drugs in Oncology (ReDO)—Propranolol as an anti-cancer agent.” ecancermedicalscience 10 (2016).
Propranolol (PRO) is a well-known and widely used non-selective beta-adrenergic receptor antagonist (beta-blocker), with a range of actions which are of interest in an oncological context. PRO displays effects on cellular proliferation and invasion, on the immune system, on the angiogenic cascade, and on tumour cell sensitivity to existing treatments. Both pre-clinical and clinical evidence of these effects, in multiple cancer types, is assessed and summarized and relevant mechanisms of action outlined. In particular there is evidence that PRO is effective at multiple points in the metastatic cascade
DCA and Celebrex COX2 inhibition Synergy
34) Li, Bo, et al. “Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells.” Oncotarget 8.31 (2017): 51748.
Dichloroacetate (DCA), a traditional mitochondria-targeting agent, has shown promising prospect as a sensitizer in fighting against malignancies including cervical cancer. But it is unclear about the effect of DCA alone on cervical tumor. Moreover, previous reports have demonstrated that the increased cyclooxygenase-2 (COX2) expression is associated with chemoresistance and poor prognosis of cervical cancer. However, it is still unknown whether COX2 can affect the sensitivity of DCA in cervical cancer cells. In this study, we found that cervical cancer cells were insensitive to DCA. Furthermore, we for the first time revealed that DCA could upregulate COX2 which impeded the chemosensitivity of DCA in cervical cancer cells. Mechanistic study showed that DCA reduced the level of RNA binding protein quaking (QKI), leading to the decay suppression of COX2 mRNA and the subsequent elevation of COX2 protein. Inhibition of COX2 using celecoxib could sensitize DCA in repressing the growth of cervical cancer cells both in vitro and in vivo. These results indicate that COX2 is a novel resistance factor of DCA, and combination of celecoxib with DCA may be beneficial to the treatment of cervical cancer.
DCA and Metformin Synergy -ETC Complex I Inhibition
35) Ward, Nathan Patrick. “Therapeutic Modulation of Cancer Metabolism with Dichloroacetate and Metformin.” (2017). Therapeutic Modulation of Cancer Metabolism with Dichloroacetate and Metformin Ward 2017
36) Ward, Nathan P., et al. “Complex I inhibition augments dichloroacetate cytotoxicity through enhancing oxidative stress in VM-M3 glioblastoma cells.” PloS one 12.6 (2017): e0180061.
The robust glycolytic metabolism of glioblastoma multiforme (GBM) has proven them susceptible to increases in oxidative metabolism induced by the pyruvate mimetic dichloroacetate (DCA). Recent reports demonstrate that the anti-diabetic drug metformin enhances the damaging oxidative stress associated with DCA treatment in cancer cells. We sought to elucidate the role of metformin’s reported activity as a mitochondrial complex I inhibitor in the enhancement of DCA cytotoxicity in VM-M3 GBM cells. Metformin potentiated DCA-induced superoxide production, which was required for enhanced cytotoxicity towards VM-M3 cells observed with the combination. Similarly, rotenone enhanced oxidative stress resultant from DCA treatment and this too was required for the noted augmentation of cytotoxicity. Adenosine monophosphate kinase (AMPK) activation was not observed with the concentration of metformin required to enhance DCA activity. Moreover, addition of an activator of AMPK did not enhance DCA cytotoxicity, whereas an inhibitor of AMPK heightened the cytotoxicity of the combination. Our data indicate that metformin enhancement of DCA cytotoxicity is dependent on complex I inhibition. Particularly, that complex I inhibition cooperates with DCA-induction of glucose oxidation to enhance cytotoxic oxidative stress in VM-M3 GBM cells.
36) Florio, Rosalba, et al. “Effects of dichloroacetate as single agent or in combination with GW6471 and metformin in paraganglioma cells.” Scientific reports 8.1 (2018): 13610.
Paragangliomas (PGLs) are infiltrating autonomic nervous system tumors that cause important morbidity. At present, surgery is the only effective therapeutic option for this rare tumor. Thus, new agents for PGL treatment should be identified. Using unique PGL cell models established in our laboratory, we evaluated the effect of dichloroacetate (DCA) as single agent or in a novel combination with other metabolic drugs, including GW6471 and metformin. DCA and metformin had not been tested before in PGL. DCA reduced PGL cell viability and growth through mechanisms involving reactivation of PDH complex leading to promotion of oxidative metabolism, with lowering of lactate and enhanced ROS production. This resulted in cell cycle inhibition and induction of apoptosis in PGL cells, as shown by flow cytometry and immunoblot analyses. Moreover, DCA drastically impaired clonogenic activity and migration of PGL cells. Also metformin reduced PGL cell viability as single agent and the combinations of DCA, GW6471 and metformin had strong effects on cell viability. Furthermore, combined treatments had drastic and synergistic effects on clonogenic ability. In conclusion, DCA, GW6471 and metformin as single agents and in combination appear to have promising antitumor effects in unique cell models of PGL.
37) Li, Bo, et al. “Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells.” Oncotarget 7.37 (2016): 59458.
Both dichloroacetate (DCA) and metformin (Met) have shown promising antitumor efficacy by regulating cancer cell metabolism. However, the DCA-mediated protective autophagy and Met-induced lactate accumulation limit their tumor-killing potential respectively. So overcoming the corresponding shortages will improve their therapeutic effects. In the present study, we found that DCA and Met synergistically inhibited the growth and enhanced the apoptosis of ovarian cancer cells. Interestingly, we for the first time revealed that Met sensitized DCA via dramatically attenuating DCA-induced Mcl-1 protein and protective autophagy, while DCA sensitized Met through markedly alleviating Met-induced excessive lactate accumulation and glucose consumption. The in vivo experiments in nude mice also showed that DCA and Met synergistically suppressed the growth of xenograft ovarian tumors. These results may pave a way for developing novel strategies for the treatment of ovarian cancer based on the combined use of DCA and Met.
38) Kolesnik, D. L., et al. “Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma.” Experimental oncology 41.1 (2019): 14-19.Cytotoxicity of Dichloroacetate and metformin against Lewis lung carcinoma Kolesnik Exper onc 2019
39) Inanc, Seniz, et al. “Metformin And Dichloroacetate Combination Exert A Synergistic Effect On Cell Viability Of Oral Squamous Cell Carcinoma.” ENT Updates 9.2 (2019): 68-73. Metformin And Dichloroacetate Synergistic Effect On Oral Squamous Cell Carcinoma Inanc Seniz ENT Updates 2019
Synergy of Metformin with Venetoclax
40) Oncotarget. 2016 Aug 9;7(32):51435-51449. doi: 10.18632/oncotarget.9843.
Biguanides sensitize leukemia cells to ABT-737-induced apoptosis by inhibiting mitochondrial electron transport. Velez J1,2, Pan R3, Lee JT4, Enciso L2, Suarez M2, Duque JE5, Jaramillo D1, Lopez C6, Morales L1, Bornmann W7, Konopleva M3, Krystal G4, Andreeff M3, Samudio I1,2,8.
Section of Molecular Hematology and Therapy, The University of Texas MD Anderson Cancer Center,
Metformin displays antileukemic effects partly due to activation of AMPK and subsequent inhibition of mTOR signaling. Nevertheless, Metformin also inhibits mitochondrial electron transport at complex I in an AMPK-independent manner, Here we report that Metformin and rotenone inhibit mitochondrial electron transport and increase triglyceride levels in leukemia cell lines, suggesting impairment of fatty acid oxidation (FAO). We also report that, like other FAO inhibitors, both agents and the related biguanide, Phenformin, increase sensitivity to apoptosis induction by the bcl-2 inhibitor ABT-737 supporting the notion that electron transport antagonizes activation of the intrinsic apoptosis pathway in leukemia cells. Both biguanides and rotenone induce superoxide generation in leukemia cells, indicating that oxidative damage may sensitize toABT-737 induced apoptosis. In addition, we demonstrate that Metformin sensitizes leukemia cells to the oligomerization of Bak, suggesting that the observed synergy with ABT-737 is mediated, at least in part, by enhanced outer mitochondrial membrane permeabilization. Notably, Phenformin was at least 10-fold more potent than Metformin in abrogating electron transport and increasing sensitivity to ABT-737, suggesting that this agent may be better suited for targeting hematological malignancies. Taken together, our results suggest that inhibition of mitochondrial metabolism by Metformin or Phenformin is associated with increased leukemia cell susceptibility to induction of intrinsic apoptosis, and provide a rationale for clinical studies exploring the efficacy of combining biguanides with the orally bioavailable derivative of ABT-737, Venetoclax.
DCA for Multiple Myeloma
40) Tian, Dandan. “Repurposing Dichloroacetate for the Treatment of Multiple Myeloma.” (2018). Repurposing Dichloroacetate for the Treatment of Multiple Myeloma Tian Dandan 2018
DCA enhances Chemotherapy
41) Lu, Xiao, et al. “Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer.” Cancer management and research 10 (2018): 1231.
DCA Salinomycin Synergy (Also Ivermectin- Stromectal ?)
42) Skeberdytė, Aistė, et al. “Dichloroacetate and salinomycin exert a synergistic cytotoxic effect in colorectal cancer cell lines.” Scientific reports 8.1 (2018): 17744.
2018 DCA and Chronic Fatigue
43) Comhaire, Frank. “Treating patients suffering from myalgic encephalopathy/chronic fatigue syndrome (ME/CFS) with sodium dichloroacetate: An open-label, proof-of-principle pilot trial.” Medical hypotheses 114 (2018): 45-48.
Twenty-two consecutive patients suffering from refractory myalgic encephalitis/chronic fatigue syndrome (ME/CFS) were treated with an innovative nutriceutical containing sodium dichloroacetate in a proof-of-principle, pilot, open-label prospective cohort trial. Ten patients experienced significant improvement of their health condition with reduction to almost half of their score in the fatigue severity scale. In twelve patients treatment failed to exert any beneficial effect. In the latter patients several other diseases have commonly been revealed by extensive biological and imaging investigations. These preliminary findings sustain the hypothetical role of mitochondrial hypo-metabolism due to inhibition of the activity of the pyruvate dehydrogenase in the pathogenesis of primary ME/CFS, and suggest a possible benefit of nutriceutical treatment by sodium dichloroacetate.
2018 Synergy of DCA combined with Curcumin Hepatoma cell line
44) Coupling Dichloroacetate Treatment with Curcumin Significantly Enhances Anticancer Potential. Kan, Ping-Chuan, et al. “Coupling dichloroacetate treatment with curcumin significantly enhances anticancer potential.” Anticancer research 38.11 (2018): 6253-6261.
Dichloroacetate (DCA) and curcumin have been shown to be potent drug candidates in cancer therapy. Our study aimed to investigate the combined effects of DCA and essential oil-blended curcumin (ECUR) using the hepatoma Huh-7 cell model.
MATERIALS AND METHODS: Muse Cell Cycle assay, Muse Annexin V & Dead Cell assay, Muse Oxidative Stress assay, and western blot analysis were applied to explore the underlying mechanisms.
Cell Cycle assay, Muse Annexin V & Dead Cell assay, Muse Oxidative Stress assay, and western blot analysis were applied to explore the underlying mechanisms.
RESULTS: DCA combined with ECUR dramatically augmented inhibition of cell survival and enhanced apoptotic induction. The enhanced apoptosis was accompanied by mitochondria-dependent apoptotic signaling activation and corroborated with significant cellular morphological alternations.
CONCLUSION: Apoptosis was the major event contributing to the synergistically boosted antiproliferative effect. Coupling DCA treatment with curcumin may rationally be expected to lower the DCA dose needed and relatively reduce accompanying toxicity and oxidative damage while enhancing anticancer potential. This novel ‘add-on’ approach is, thus, of enormous value to the current DCA therapy.
2014 DCA resensitized Gastric CA cells to Chemo (5FU)
45) Xuan, Yi, et al. “Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism.” Experimental cell research 321.2 (2014): 219-230.Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism.
In this study, we investigated whether gastric cancer with hypoxia-induced resistance to 5-fluorouracil (5-FU) could be re-sensitized following treatment with low-dose dichloroacetate (DCA), an inhibitor of the glycolytic pathway. The expression profiles of hypoxia-inducible factor-1α (HIF-1α) and pyruvate dehydrogenase kinase-1 (PDK-1) were analyzed in tissues from 10 patients with gastric cancer who had different responses to adjuvant 5-FU treatment. For the in vitro assays, cell viability and apoptosis were evaluated with and without treatment with 20mM DCA in the AGS and MKN45 cell lines, as well as in PDK1 knockdown cell lines. The expression levels of HIF-1α and PDK-1 were both elevated in the tumor tissues relative to the normal gastric tissues of most patients who showed recurrence after adjuvant 5-FU treatment. Cellular viability tests showed that these cell lines had a lower sensitivity to 5-FU under hypoxic conditions compared to normoxic conditions. Moreover, the addition of 20mM DCA only increased the sensitivity of these cells to 5-FU under hypoxic conditions, and the resistance to 5-FU under hypoxia was also attenuated in PDK1 knockdown cell lines. In conclusion, DCA treatment was able to re-sensitize gastric cancer cells with hypoxia-induced resistance to 5-FU through the alteration of glucose metabolism.
—————— ————————— —————–
Poly MVA and DCA – Paul Anderson
These files Courtesy of and Copyright (c) Paul S. Anderson ND
46) DCA-PolyMVA-Protocol Paul Anderson 2017
Combined Protocol of DCA with PolyMVA (LAMC): In 2009 as part of a “non-responder” arm of an NIH funded trial we (7) surmised a potential synergy of LAMC and DCA. A cell line study (1,8) showed apoptotic cell synergy in GBM cells. Additionally chemically the two agents have a potential for physiologic mutual benefit.
47) Metabolic Therapies in Advanced “Salvage” Cancer Cases
by Dr. Paul S. Anderson, NMD Townsend Letter Aug/Sept 2018
An excellent example of true therapeutic synergy was discovered in the earlier days of my IV research (under the NIH-funded Bastyr Integrative Oncology Research Center) in the combination therapy using Poly-MVA and dichloroacetate (DCA) for both IV and oral use. We will describe the basis of the synergy and the initial case series that was followed.
48) Dr Paul Anderson book chapter on DCA combined with poly-MVA (alpha lipoic acid)
49) Rationale for and Protocol for the use of combined Dichloroacetate (DCA) and Lipoic Acid Mineral Complex (LAMC) in advanced Cancer Patients as developed by Paul S. Anderson for patients treated at Anderson Medical Specialty Associates and in the Bastyr University Clinical Research Center (BCRC) Dichloroacetate (DCA) and Lipoic Acid Mineral Complex (LAMC) PolyMVA in advanced Cancer Patients Paul S. Anderson
50) Dr Paul Anderson book chapter on DCA combined with poly-MVA (alpha lipoic acid) Outside-the-Box-Cancer-Therapies-Paul Anderson _Mark-Stengler
—————— ——————————–
PolyMVA for Cancer Sheri Lieberman Aug 2008
51) Poly-MVA as an integrative approach to the treatment of cancer: evidence-based through case reports. Sheri Lieberman Aug 2008 Townsend letter.
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
DCA has immunomodulatory activity, toward T1 function
53) Badr, Mujtaba M., et al. “Dichloroacetate modulates cytokines toward T helper 1 function via induction of the interleukin-12–interferon-gamma pathway.” OncoTargets and therapy 7 (2014): 193.
DCA has immunomodulatory activity, mainly via activation of the IL-12–IFN-gamma pathway and is able to modulate cytokines toward T helper 1 lymphocyte function. These DCA immunomodulatory effects are promising and further investigations are required to develop protocols for its use in cancer treatment.
————————– —————————– —————
54) D’s Lymphoma Remission DCASITE
Email, 5 September 2008. Dear Jim, I have attached a pdf file that contains my 3 and 6 month PET-CAT scan results. …I have gotten some extremely good results. … I hope you like the info in the PDF. I certainly do! Best regards,D Below are scans and a copy of the reports. And following that is a detailed email describing the situation.
———————————— ————————————-
55) Case Reports LEMMO Integrated Cancer Care Inc has been pioneering the use of DCA by intravenous injection and oral routes since 2008.
—————————————————
DCA and Artesunate Toxicity – Caused Liver Failure and Death
56) 74) Uhl, Martin, Stefan Schwab, and Thomas Efferth. “Fatal liver and bone marrow toxicity by combination treatment of dichloroacetate and artesunate in a glioblastoma multiforme patient: case report and review of the literature.” Frontiers in oncology 6 (2016): 204.
57) 75) Brandsma, Dieta, et al. “Severe encephalopathy and polyneuropathy induced by dichloroacetate.” Journal of neurology 257.12 (2010): 2099-2100.Severe encephalopathy and polyneuropathy induced by dichloroacetate Brandsma J neurology 2010
——————————– ————————– ————–
DCA Inhibits Neuroblastoma
58) 76) Vella, Serena, et al. “Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells.” International journal of cancer 130.7 (2012): 1484-1493.Dichloroacetate inhibits neuroblastoma growth against malignant undifferentiated cells Vella Serena Int j cancer 2012
Combine DCA with Phenylbutyrate Synergy
59) 77) Ferriero, Rosa, et al. “Differential inhibition of PDKs by phenylbutyrate and enhancement of pyruvate dehydrogenase complex activity by combination with dichloroacetate.” Journal of Inherited Metabolic Disease 38.5 (2015): 895.
Pyruvate dehydrogenase complex (PDHC) is a key enzyme in metabolism linking glycolysis to tricarboxylic acid cycle and its activity is tightly regulated by phosphorylation catalyzed by four pyruvate dehydrogenase kinase (PDK) isoforms. PDKs are pharmacological targets for several human diseases including cancer, diabetes, obesity, heart failure, and inherited PDHC deficiency. We investigated the inhibitory activity of phenylbutyrate toward PDKs and found that PDK isoforms 1-to-3 are inhibited whereas PDK4 is unaffected. Moreover, docking studies revealed putative binding sites of phenylbutyrate on PDK2 and 3 that are located on different sites compared to dichloroacetate (DCA), a previously known PDK inhibitor. Based on these findings, we showed both in cells and in mice that phenylbutyrate combined to DCA results in greater increase of PDHC activity compared to each drug alone. These results suggest that therapeutic efficacy can be enhanced by combination of drugs increasing PDHC enzyme activity.
——————- ———————–
60) 78) Cancer Treatment Research Page DCA Info PAge
==================================
DCA Induces Regulatory T Cell Differentiation-This could result in decreased immunosurveillance in case of its use as an anticancer drug.
61) 79) Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Antoniadis N, Liakopoulos V, Stefanidis I. J Basic Clin Physiol Pharmacol. 2013;24(4):271-6.
Most cancer cells rely on aerobic glycolysis. Dichloroacetate (DCA) inhibits aerobic glycolysis and is a promising relatively nontoxic anticancer compound. However, rapidly proliferating effector T-cells also rely on aerobic glycolysis, whereas regulatory T-cells (Treg) do not. The effect of DCA on glucose metabolism and Treg differentiation was evaluated in alloreactive lymphocytes.
METHODS:Peripheral blood mononuclear cells from healthy volunteers were used in a two-way mixed lymphocyte reaction. Lymphocyte proliferation was assessed by cell counting; DCA cytotoxicity, by lactate dehydrogenase release assay; and glucose uptake and aerobic glycolysis, by measuring in the supernatants the correspondent glucose and lactate concentrations. Interleukin-10 (IL-10) was measured in the supernatants, whereas the Treg signature transcription factor forkhead box P3 (FOXP3) was measured in cell lysates by means of enzyme-linked immunosorbent assay.
RESULTS:DCA had a minor effect on lymphocyte proliferation and cytotoxicity. However, DCA decreased glucose uptake and inhibited aerobic glycolysis. Finally, DCA markedly increased the production of IL-10 and the expression of FOXP3.
CONCLUSIONS:DCA inhibits aerobic glycolysis and induces Treg differentiation in human alloreactive lymphocytes. This could result in decreased immunosurveillance in case of its use as an anticancer drug. However, DCA could play a role as an immunosuppressant in the fields of transplantation and autoimmunity.
=================== ==========
62) 80) Stakišaitis, Donatas, et al. “The Importance of Gender-Related Anticancer Research on Mitochondrial Regulator Sodium Dichloroacetate in Preclinical Studies In Vivo.” Cancers 11.8 (2019): 1210.
63) 81) Kankotia, Shyam, and Peter W. Stacpoole. “Dichloroacetate and cancer: new home for an orphan drug?.” Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 1846.2 (2014): 617-629.
We reviewed the anti-cancer effects of DCA, an orphan drug long used as an investigational treatment for various acquired and congenital disorders of mitochondrial intermediary metabolism. Inhibition by DCA of mitochondrial pyruvate dehydrogenase kinases and subsequent reactivation of the pyruvate dehydrogenase complex and oxidative phosphorylation is the common mechanism accounting for the drug’s anti-neoplastic effects. At least two fundamental changes in tumor metabolism are induced by DCA that antagonize tumor growth, metastases and survival: the first is the redirection of glucose metabolism from glycolysis to oxidation (reversal of the Warburg effect), leading to inhibition of proliferation and induction of caspase-mediated apoptosis. These effects have been replicated in both human cancer cell lines and in tumor implants of diverse germ line origin. The second fundamental change is the oxidative removal of lactate, via pyruvate, and the co-incident buffering of hydrogen ions by dehydrogenases located in the mitochondrial matrix. Preclinical studies demonstrate that DCA has additive or synergistic effects when used in combination with standard agents designed to modify tumor oxidative stress, vascular remodeling, DNA integrity or immunity. These findings and limited clinical results suggest that potentially fruitful areas for additional clinical trials include 1) adult and pediatric high grade astrocytomas; 2) BRAF-mutant cancers, such as melanoma, perhaps combined with other pro-oxidants; 3) tumors in which resistance to standard platinum-class drugs alone may be overcome with combination therapy; and 4) tumors of endodermal origin (GI Lung), in which extensive experimental research has demonstrated significant anti-proliferative, pro-apoptotic effects of DCA, leading to improved host survival.
======================================================
DCA dichloroacetate for leukemia, lymphoma and myeloma.
64) 82) Villalba, Martin, et al. “Chemical metabolic inhibitors for the treatment of blood-borne cancers.” Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) 14.2 (2014): 223-232.
One enzyme implicated in tumor metabolic remodeling and whose expression is regulated by oncogenic transcription factors is pyruvate dehydrogenase kinase 1 (PDK1), [3]), which is inhibited by dichloroacetate (DCA). PDK1 inhibition leads to pyruvate dehydrogenase (PDH) activation and forces cells to use mitochondria as the main ATP generator. As a result, glycolysis is vastly diminished.
=======================
DCA for Squamous Cell CA
65) 83) Ruggieri, Vitalba, et al. “Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment.” Oncotarget 6.2 (2015): 1217.
Reprogramming of metabolism is a well-established property of cancer cells that is receiving growing attention as potential therapeutic target. Oral squamous cell carcinomas (OSCC) are aggressive and drugs-resistant human tumours displaying wide metabolic heterogeneity depending on their malignant genotype and stage of development. Dichloroacetate (DCA) is a specific inhibitor of the PDH-regulator PDK proved to foster mitochondrial oxidation of pyruvate. In this study we tested comparatively the effects of DCA on three different OSCC-derived cell lines, HSC-2, HSC-3, PE15. Characterization of the three cell lines unveiled for HSC-2 and HSC-3 a glycolysis-reliant metabolism whereas PE15 accomplished an efficient mitochondrial oxidative phosphorylation. DCA treatment of the three OSCC cell lines, at pharmacological concentrations, resulted in stimulation of the respiratory activity and caused a remarkably distinctive pro-apoptotic/cytostatic effect on HSC-2 and HSC-3. This was accompanied with a large remodeling of the mitochondrial network, never documented before, leading to organelle fragmentation and with enhanced production of reactive oxygen species. The data here presented indicate that the therapeutic efficacy of DCA may depend on the specific metabolic profile adopted by the cancer cells with those exhibiting a deficient mitochondrial oxidative phosphorylation resulting more sensitive to the drug treatment.
==========================================
Dr Akbar Khan Medicor Clinic Toronto
66) 84) DCA Case Studies from Medicor Clinic Toronto
Case Studies DCA in Cancer Patients
- Metastatic Renal Carcinoma
- Metastatic Lung Carcinoma
- Metastatic Ovarian Carcinoma – Case 1
- Ampulla of Vater Carcinoma
- Metastatic Ovarian Carcinoma – Case 2
- Glioblastoma Multiforme – Case 1
- Mesothelioma
- Melanoma with Brain Metastases
- Metastatic Ovarian Cancer
- Glioblastoma Multiforme – Case 2
==========================
Dr Akbar Khan Case reports Medicor Clinic Toronto
67) 85) Khan, Akbar, et al. “A novel form of dichloroacetate therapy for patients with advanced cancer: a report of 3 cases.” Alternative Therapies in Health & Medicine 20 (2014).Dichloroacetate DCA for advanced cancer report of 3 cases Khan, Akbar Alternative Therapies in Health & Medicine 2014
68) 86) Khan, Akbar, Douglas Andrews, and Anneke C. Blackburn. “Long-term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy.” World journal of clinical cases 4.10 (2016): 336.
Oral dichloroacetate sodium (DCA) has been investigated as a novel metabolic therapy for various cancers since 2007, based on data from Bonnet et al that DCA can trigger apoptosis of human lung, breast and brain cancer cells. Response to therapy in human studies is measured by standard RECIST definitions, which define “response” by the degree of tumour reduction, or tumour disappearance on imaging. However, Blackburn et al have demonstrated that DCA can also act as a cytostatic agent in vitro and in vivo, without causing apoptosis (programmed cell death). A case is presented in which oral DCA therapy resulted in tumour stabilization of stage 4 colon cancer in a 57 years old female for a period of nearly 4 years, with no serious toxicity. Since the natural history of stage 4 colon cancer consists of steady progression leading to disability and death, this case highlights a novel use of DCA as a cytostatic agent with a potential to maintain long-term stability of advanced-stage cancer.
Keywords: Dichloroacetate, Cancer, Colon, Colorectal, Cytostatic, Stabilization, Growth inhibition, Intravenous
Core tip: Oral dichloroacetate sodium (DCA) has been investigated as a novel metabolic therapy for various cancers. Response to therapy in human studies is measured by standard RECIST definitions, which define “response” by the degree of tumour reduction, or tumour disappearance on imaging. However, DCA can also act as a cytostatic agent, without causing apoptosis (programmed cell death). A case is presented in which oral DCA therapy resulted in tumour stabilization of stage 4 colon cancer in a 57 years old female for a period of nearly 4 years, with no serious toxicity.
The oral DCA regimen that was developed included three natural medications acetyl L-carnitine[29-31], R-alpha lipoic acid[32-34] and benfotiamine[35-37], for the primary purpose of neuropathy prevention.
Observational data collected from more than 300 cancer patients with advanced disease revealed measurable benefits from DCA therapy in 60%-70% of cases. The neuropathy risk with inclusion of natural neuroprotective agents was roughly 20% with 20-25 mg/kg per day dosing on a 2 wk on/1 wk off cycle. Reversible liver enzyme elevation was noted in approximately 2% in this patient group (clinic observational data published online at
Chu et al[11] reported on 24 patients treated for a median time of 2 mo at either 6.25 or 12.5 mg/kg BID, on continuous oral DCA without neuroprotective supplements. They concluded that the recommended phase 2 dose was 6.25 mg/kg BID (12.5 mg/kg per day), with careful monitoring of neuropathy being needed. Dunbar et al[9] recommended 5 mg/kg BID as a starting dose for most patients, with their trial administering 4, 8 or 12.5 mg/kg BID continuously (median time on DCA 34 d), also without neuroprotective supplements. The patient in this report took 500 mg BID, equivalent to 8.2 mg/kg BID, 2 wk on/1 wk off, but could not tolerate this dose three times a day (total of 25 mg/kg per day)
69) 87) Khan, Akbar, et al. “Long-term stabilization of metastatic melanoma with sodium dichloroacetate.” World journal of clinical oncology 8.4 (2017): 371.Correspondence to: Akbar Khan, MD, Medical Director, Medicor Cancer Centres Inc, 4576 Yonge St., Suite 301, Toronto, ON M2N 6N4, Canada.
Sodium dichloroacetate (DCA) has been studied as a metabolic cancer therapy since 2007, based on a publication from Bonnet et al demonstrating that DCA can induce apoptosis (programmed cell death) in human breast, lung and brain cancer cells. Classically, the response of cancer to a medical therapy in human research is measured by Response Evaluation Criterial for Solid Tumours definitions, which define “response” by the degree of tumour reduction, or tumour disappearance on imaging, however disease stabilization is also a beneficial clinical outcome. It has been shown that DCA can function as a cytostatic agent in vitro and in vivo, without causing apoptosis. A case of a 32-year-old male is presented in which DCA therapy, with no concurrent conventional therapy, resulted in regression and stabilization of recurrent metastatic melanoma for over 4 years’ duration, with trivial side effects. This case demonstrates that DCA can be used to reduce disease volume and maintain long-term stability in patients with advanced melanoma.
================= ==================
Dr Akbar Khan Medicor Clinic Toronto
Medicor Cancer Centres
Medicor Cancer Centres
4576 Yonge St, Suite 301
Toronto, ON Canada M2N 6N4
Tel: +1-416-227-0037
Fax: +1-416-227-1915
Staff Emails
For General Information: info@medicorcancer.com
Office Manager – Yasmine: yibrahim@medicorcancer.com
Logistics Manager – Maggie: mdelaney@medicorcancer.com
Director of Naturopathic Medicine – Dr. Doug Andrews: dandrews@medicorcancer.com
Medical Director – Dr. Akbar Khan: akhan@medicorcancer.com
President – Dr. Humaira Khan: hkhan@medicorcancer.com
70) Khan, Akbar, et al. “Long-term stabilization of metastatic melanoma with sodium dichloroacetate.” World journal of clinical oncology 8.4 (2017): 371.
Sodium dichloroacetate (DCA) has been studied as a metabolic cancer therapy since 2007, based on a publication from Bonnet et al demonstrating that DCA can induce apoptosis (programmed cell death) in human breast, lung and brain cancer cells. Classically, the response of cancer to a medical therapy in human research is measured by Response Evaluation Criterial for Solid Tumours definitions, which define “response” by the degree of tumour reduction, or tumour disappearance on imaging, however disease stabilization is also a beneficial clinical outcome. It has been shown that DCA can function as a cytostatic agent in vitro and in vivo, without causing apoptosis. A case of a 32-year-old male is presented in which DCA therapy, with no concurrent conventional therapy, resulted in regression and stabilization of recurrent metastatic melanoma for over 4 years’ duration, with trivial side effects. This case demonstrates that DCA can be used to reduce disease volume and maintain long-term stability in patients with advanced melanoma.
89) Khan, Akbar, Douglas Andrews, and Anneke C. Blackburn. “Long-term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy.” World journal of clinical cases 4.10 (2016): 336.
DCA and Celebrex Synergy in Cervical CA
71) 90) Li, Bo, et al. “Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells.” Oncotarget 8.31 (2017): 51748. Dichloroacetate (DCA), a traditional mitochondria-targeting agent, has shown promising prospect as a sensitizer in fighting against malignancies including cervical cancer. But it is unclear about the effect of DCA alone on cervical tumor. Moreover, previous reports have demonstrated that the increased cyclooxygenase-2 (COX2) expression is associated with chemoresistance and poor prognosis of cervical cancer. However, it is still unknown whether COX2 can affect the sensitivity of DCA in cervical cancer cells. In this study, we found that cervical cancer cells were insensitive to DCA. Furthermore, we for the first time revealed that DCA could upregulate COX2 which impeded the chemosensitivity of DCA in cervical cancer cells. Mechanistic study showed that DCA reduced the level of RNA binding protein quaking (QKI), leading to the decay suppression of COX2 mRNA and the subsequent elevation of COX2 protein. Inhibition of COX2 using celecoxib could sensitize DCA in repressing the growth of cervical cancer cells both in vitro and in vivo. These results indicate that COX2 is a novel resistance factor of DCA, and combination of celecoxib with DCA may be beneficial to the treatment of cervical cancer.
DCA Adverse Effects Toxicity
72A) 91) Sen, R., et al. “LONG-TERM ADMINISTRATION OF DCA INDUCES EPIDIDYMIS TOXICITY IN MALE ALBINO RATS.” Advances in Pharmacology and Toxicology 17.1 (2016): 21.
72B) A TOXICOLOGICAL STUDY OF DICHLOROACETIC ACID (DCA) ON THE PROSTATE GLAND OF MALE ALBINO RATS. Source: Advances in Pharmacology & Toxicology . Dec2015, Vol. 16 Issue 3, p2-17. 7p. Author(s): Sen, R.; Shaffi, S. A.; Kakaria, V. K.; Chauhan, A.
Abstract: Dichloroacetic acid (DCA) is chlorinated disinfection by-product (CDBPs) and also used as a therapeutic agent. It is formed as a result of chlorination in drinking water (At that time when acetic acid in which two of the three hydrogen atoms of the methyl group have been replaced by the chlorine atoms in drinking water then DCA form). In the present study, male albino rats were orally administered with 125mg/kg-body weight of Dichloroacetate for 30 days, 60 days and 90 days. The animals were sacrificed and prostate gland was quickly dissected out and fix in 10% formalin for rutine histological techniques. The 30 days treated rats, histology showed a slight change in the structure of acini, all epithelial layers shrinkage and the acini and fibro muscular tissues affected at 60 days and all epithelial layers shrinkage, and clump in glandular ducts and fibro muscular tissue affected at 90 days. Pathological changes seen in the prostate gland has been correlated with the possible alteration in the gonadal hormone. It is suggested that DCA is probable prostate carcinogenic and reproductive toxicant in rats. Adverse effects of DCA include polyneuropathy and testicular degeneration.
Copyright of Advances in Pharmacology & Toxicology is the property of Advances in Pharmacology & Toxicology and its content may not be copied or emailed to multiple sites or posted to a listserv without the copyright holder’s express written permission. However, users may print, download, or email articles for individual use. This abstract may be abridged. No warranty is given about the accuracy of the copy. Users should refer to the original published version of the material for the full abstract.
Fenofibrate separates HK2 from VDAC
73) 93) Int J Biol Sci. 2016; 12(7): 786–798.
Fenofibrate Suppresses Oral Tumorigenesis via Reprogramming Metabolic Processes: Potential Drug Repurposing for Oral Cancer
Chia-Ing Jan,1,2 Ming-Hsui Tsai,3 Chang-Fang Chiu,4 Yi-Ping Huang,5 Chia Jen Liu,6 and Nai Wen Chang
Fenofibrate induced cytotoxicity by decreasing oxygen consumption rate (OCR) that was accompanied with increasing extracellular acidification rate (ECAR) and reducing ATP content. Moreover, fenofibrate caused changes in the protein expressions of hexokinase II (HK II), pyruvate kinase, pyruvate dehydrogenase, and voltage-dependent anion channel (VDAC), which are associated with the Warburg effect. In addition, fenofibrate reprogrammed the metabolic pathway by interrupting the binding of HK II to VDAC.
One anticancer strategy suggests targeting mitochondrial metabolism to trigger cell death through slowing down energy production from the Warburg effect. Fenofibrate is a clinical lipid-lowering agent and an effective anticancer drug. In the present study, we demonstrate that fenofibrate provided novel mechanisms for delaying oral tumor development via the reprogramming of metabolic processes. Fenofibrate induced cytotoxicity by decreasing oxygen consumption rate (OCR) that was accompanied with increasing extracellular acidification rate (ECAR) and reducing ATP content. Moreover, fenofibrate caused changes in the protein expressions of hexokinase II (HK II), pyruvate kinase, pyruvate dehydrogenase, and voltage-dependent anion channel (VDAC), which are associated with the Warburg effect. In addition, fenofibrate reprogrammed the metabolic pathway by interrupting the binding of HK II to VDAC. In an oral cancer mouse model, fenofibrate exhibited both preventive and therapeutic efficacy on oral tumorigenesis. Fenofibrate administration suppressed the incidence rate of tongue lesions, reduced the tumor sizes, decreased the tumor multiplicity, and decreased the immunoreactivities of VDAC and mTOR. The molecular mechanisms involved in fenofibrate’s ability to delay tumor development included the down-regulation of mTOR activity via TSC1/2-dependent signaling through activation of AMPK and inactivation of Akt, or via a TSC1/2-independent pathway through direct suppression of raptor. Our findings provide a molecular rationale whereby fenofibrate exerts anticancer and additional beneficial effects for the treatment of oral cancer patients.
Clotrimazole Detaches HK2 from Mitochondria
CLotrimazole coordination with Palladium transition Metal
Excellent Review !!!!!!
74) 94) Kadavakollu, S., et al. “Clotrimazole as a cancer drug: a short review.” Medicinal chemistry 4.11 (2014): 722.
Clotrimazole disrupts HK binding to mitochondria thus precipitating cell death [14]. HK is not the only cellular target affected by clotrimazole; it also affects the two other important glycolytic enzymes: phosphofructokinase (PFK) and aldolase (ALD)
The nitrogen atom present in the imidazole ring of clotrimazole facilitates coordination with transition metal ions such as Pt, as well as Ru, Pd, Cu, Co, Zn and Ni. these metal complexes are inducing apoptosis superior to that of either the parent clotrimazole or cisplatin.
Anti-cancer effects can also be traced to its targeting of the glycolytic enzymes hexokinase, phosphofructokinase, and aldolase [12-14].
75) 95) Penso, Julia, and Rivka Beitner. “Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells.” European journal of pharmacology 342.1 (1998): 113-117.
Cancer cells are characterized by a high rate of glycolysis. Hexokinase (ATP: D-hexose 6-phosphotransferase, EC 2.7.1.1), the only glycolytic enzyme which binds to mitochondria, is exceptionally high in cancer cells, and believed to play a key role in regulating cell energy metabolism and cancer cell growth rate. We have previously found that clotrimazole (1-(alpha-2-chlorotrityl)imidazole) and bifonazole (1-(alpha-biphenyl-4-ylbenzyl)imidazole), the antifungal azole derivatives, which were recently recognized as calmodulin antagonists, are calmodulin antagonists which most effectively reduce glycolysis and ATP level in B16 melanoma cells. They act through allosteric regulation and detachment of glycolytic enzymes from cytoskeleton. Here we report of a novel, additional, mechanism of action of these drugs. We show that they induce a dose-dependent detachment of hexokinase from mitochondria of B16 melanoma cells. This effect preceded the decrease in cell viability. These results suggest that clotrimazole and bifonazole may be promising drugs in treatment of melanoma.
Lithium Detaches HK2 from Mitochondria
76) 96) Penso J1, Beitner R.Lithium detaches hexokinase from mitochondria and inhibits proliferation of B16 melanoma cells.Mol Genet Metab. 2003 Jan;78(1):74-8. Health Sciences Research Center, Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan 52900, Israel.
Glycolysis is known to be the primary energy source in cancer cells. Hexokinase (ATP: D-hexose 6-phosphotransferase, EC 2.7.1.1), the only glycolytic enzyme which binds to mitochondria, is exceptionally high in cancer cells, and believed to play a key role in regulating cell energy metabolism and cancer cell growth rate. We show here that lithium induces a detachment of hexokinase from mitochondria of B16 melanoma cells. This effect eventually leads to inhibition of cell proliferation. These results reveal a novel, additional, mechanism of action of lithium and suggest that lithium may be promising drug in treatment of melanoma.
Methyl Jasmonate
77) 97) Goldin N1, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, Bronner V, Notcovich A, Shoshan-Barmatz V, Flescher E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene. 2008 Aug 7;27(34):4636-43. Department of Human Microbiology, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel.
Cellular bio-energetic metabolism and mitochondria are recognized as potential targets for anticancer agents, due to the numerous relevant peculiarities cancer cells exhibit. Jasmonates are anticancer agents that interact directly with mitochondria. The aim of this study was to identify mitochondrial molecular targets of jasmonates. We report that jasmonates bind to hexokinase and detach it from the mitochondria and its mitochondrial anchor-the voltage-dependent anion channel (VDAC), as judged by hexokinase immunochemical and activity determinations, surface plasmon resonance analysis and planar lipid bilayer VDAC-activity analysis. Furthermore, the susceptibility of cancer cells and mitochondria to jasmonates is dependent on the expression of hexokinase, evaluated using hexokinase-overexpressing transfectants and its mitochondrial association. Many types of cancer cells exhibit overexpression of the key glycolytic enzyme, hexokinase, and its excessive binding to mitochondria. These characteristics are considered to play a pivotal role in cancer cell growth rate and survival. Thus, our findings provide an explanation for the selective effects of jasmonates on cancer cells. Most importantly, this is the first demonstration of a cytotoxic mechanism based on direct interaction between an anticancer agent and hexokinase. The proposed mechanism can serve to guide development of a new selective approach for cancer therapy.
78) 98) Leukemia. 2002 Apr;16(4):608-16.Plant stress hormones suppress the proliferation and induce apoptosis in human cancer cells.Fingrut O1, Flescher E. Department of Human Microbiology, Sackler Faculty of Medicine, Tel-Aviv University, Tel Aviv, Israel.
Cellular stressors induce various outcomes including inhibition of cell proliferation and cell death. Sodium salicylate (SA), a plant stress hormone, can suppress the proliferation or cause apoptosis in certain mammalian cancer cells. Plant stress hormones are activators of cellular responses, including cell death, to diverse stress situations in plants. Thus, we hypothesized that plant stress hormones share the ability to adversely affect cancer cells. We found that the plant stress hormone SA suppressed proliferation of lymphoblastic leukemia, prostate, breast and melanoma human cancer cells. Jasmonic acid (JA), a plant stress hormone belonging to the Jasmonate family, induced death in lymphoblastic leukemia cells and caused suppression of cell proliferation in the other human cancer cells mentioned above. Another member of the Jasmonate family, methyl jasmonate (MJ), induced death in each of the cell lines. Plant stress hormones did not affect normal human lymphocytes, in contrast to their strong effect on lymphoblastic leukemia cells. JA and MJ caused apoptotic death, as determined by characteristic nuclear morphology, flow cytometric DNA profile and elevation of caspase-3 activity. Finally, mice bearing EL-4 lymphoma and treated with MJ, survived for significantly (P = 0.00953) longer periods of time than untreated mice. These findings suggest that plant stress hormones may potentially be a novel class of anti-cancer drugs.
79) 99) Fingrut O1, Reischer D, Rotem R, Goldin N, Altboum I, Zan-Bar I, Flescher E. Jasmonates induce nonapoptotic death in high-resistance mutant p53-expressing B-lymphoma cells. Br J Pharmacol. 2005 Nov;146(6):800-8.Department of Human Microbiology, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv 69978, Israel.
Interestingly, methyl jasmonate was not toxic towards human sperm cells in vitro (Flescher, personal communication). The relative independence of sperm cells from oxidative phosphorylation as a supplier of ATP may account for the insensitivity of these cells to methyl jasmonate.
Jasmonate-induced death of B-lymphoma cells involves severe ATP depletion.
80) 100) Sucu, Bilgesu Onur, et al. “Synthesis of novel methyl jasmonate derivatives and evaluation of their biological activity in various cancer cell lines.” Bioorganic chemistry 91 (2019): 103146.
2017 Nice Diagram !!!!
81) 101) Li, Jingjing, et al. “Methyl jasmonate leads to necrosis and apoptosis in hepatocellular carcinoma cells via inhibition of glycolysis and represses tumor growth in mice.” Oncotarget 8.28 (2017): 45965.
Methyl jasmonate has recently been found to have anti-cancer activity. Methyl jasmonate detached hexokinase 2 from a voltage dependent anion channel causing a reduction in mitochondrial transmembrane potential that led to the release of cytochrome C and apoptosis inducing factor resulting in intrinsic apoptosis. Blocked adenosine triphosphate synthesis caused by mitochondrial injury hampered oxidative phosphorylation and led to cell necrosis. The results were applied to the in vivo treatment of nude mice with a satisfactory effect. Collectively, our results suggest that methyl jasmonate may be an adjuvant therapy for liver tumors due to its mechanism in cancer cells compared to that in normal cells: The major function is to inhibit glycolysis instead of changing aerobic metabolism.
the major effect of methyl jasmonate in HCC cells was profoundly different to its effect on normal cells: methyl jasmonate interfered with the growth of cancer cells by inhibiting glycolysis. The underlying mechanism was dependent on necrosis and apoptosis by detaching HK2 from VDAC1 causing loss of mitochondrial function.
82) 102) Wang, Yao, et al. “EZH2 inhibition promotes methyl jasmonate‑induced apoptosis of human colorectal cancer through the Wnt/β‑catenin pathway.” Oncology letters 16.1 (2018): 1231-1236.
83) 103) Yehia, Rania, et al. “Anti-tumor efficacy of an integrated methyl dihydrojasmonate transdermal microemulsion system targeting breast cancer cells: in vitro and in vivo studies.” Colloids and Surfaces B: Biointerfaces 155 (2017): 512-521.
84) 104) Zhang, Mutian, et al. “Methyl jasmonate induces apoptosis and pro-apoptotic autophagy via the ROS pathway in human non-small cell lung cancer.” American journal of cancer research 6.2 (2016): 187.
85) 105) Cesari, Italo Mario, et al. “Methyl jasmonate: putative mechanisms of action on cancer cells cycle, metabolism, and apoptosis.” International journal of cell biology 2014 (2014).
86) 106) Tong, Qiang-Song, et al. “Methyl jasmonate downregulates expression of proliferating cell nuclear antigen and induces apoptosis in human neuroblastoma cell lines.” Anti-Cancer Drugs 19.6 (2008): 573-581.
Cellular stressors induce various outcomes including inhibition of cell proliferation and cell death. Sodium salicylate (SA), a plant stress hormone, can suppress the proliferation or cause apoptosis in certain mammalian cancer cells. Plant stress hormones are activators of cellular responses, including cell death, to diverse stress situations in plants. Thus, we hypothesized that plant stress hormones share the ability to adversely affect cancer cells. We found that the plant stress hormone SA suppressed proliferation of lymphoblastic leukemia, prostate, breast and melanoma human cancer cells. Jasmonic acid (JA), a plant stress hormone belonging to the Jasmonate family, induced death in lymphoblastic leukemia cells and caused suppression of cell proliferation in the other human cancer cells mentioned above. Another member of the Jasmonate family, methyl jasmonate (MJ), induced death in each of the cell lines. Plant stress hormones did not affect normal human lymphocytes, in contrast to their strong effect on lymphoblastic leukemia cells. JA and MJ caused apoptotic death, as determined by characteristic nuclear morphology, flow cytometric DNA profile and elevation of caspase-3 activity. Finally, mice bearing EL-4 lymphoma and treated with MJ, survived for significantly (P = 0.00953) longer periods of time than untreated mice. These findings suggest that plant stress hormones may potentially be a novel class of anti-cancer drugs.
88) 108) Kim JH, Lee SY, Oh, SY, Han SI, Park HG, Yoo M, Kang HS. Methyl jasmonate induces apoptosis through induction of Bax/Bcl-XS and activation of caspase-3 via ROS production in A549 cells. Oncol Rep 2004; 12:1233-8;
89) Rotem R, Heyfets A, Fingrut O, Blickstein D, Shaklai M, Flescher E. Jasmonates: novel anticancer agents acting directly and selectively on human cancer cell mitochondria. Canc Res 2005; 65:1981-993;
90) 109) https://www.researchgate.net/profile/Caroline_Picoli/publication/320641189_Methyl_jasmonate_A_phytohormone_with_potential_for_the_treatment_of_inflammatory_bowel_diseases/links/5b3b779a4585150d23f2fff4/Methyl-jasmonate-A-phytohormone-with-potential-for-the-treatment-of-inflammatory-bowel-diseases.pdf
MeJA: a potential phytohormone foranti-TNF therapy in the treatment of IBDs
91) 110) Besson, Jean Carlos Fernando, et al. “Methyl jasmonate: a phytohormone with potential for the treatment of inflammatory bowel diseases.” Journal of Pharmacy and Pharmacology 70.2 (2018): 178-190.
92) 111) Gunjegaonkar, S. M., and T. S. Shanmugarajan. “Molecular mechanism of plant stress hormone methyl jasmonate for its anti-inflammatory activity.” Plant signaling & behavior 14.10 (2019): e1642038.
——————————
Methyl Jasmonate from TCI
93) 112)
Methyl Jasmonate (mixture of isomers) Tokyo Chemical Industry
(CAS RN:1101843-02-0 Product Number:M1068)
——————————– —
Metformin – Complex I of ETC
94) 113) Andrzejewski, Sylvia, et al. “Metformin directly acts on mitochondria to alter cellular bioenergetics.” Cancer & metabolism 2.1 (2014): 12.
95) 114) Vial, Guillaume, Dominique Detaille, and Bruno Guigas. “Role of Mitochondria in the Mechanism (s) of Action of Metformin.” Frontiers in endocrinology 10 (2019): 294. Inhibition of Complex 1 by Metformin and Metabolic Reprogramming in Cancer Cells
96) Stacpoole, Peter W., et al. “Dichloroacetate-induced peripheral neuropathy.” International review of neurobiology 145 (2019): 211-238.
Pterostilbene for Mantle Cell Lymphoma PI3K/Akt/mTOR
97) 115) Yu, Dandan, et al. “Targeting the PI3K/Akt/mTOR signaling pathway by pterostilbene attenuates mantle cell lymphoma progression.” Acta biochimica et biophysica Sinica 50.8 (2018): 782-792.
Mantle cell lymphoma (MCL) is an aggressive and mostly incurable B-cell malignancy with frequent relapses after an initial response to standard chemotherapy. Therefore, novel therapies are urgently required to improve MCL clinical outcomes. In this study, MCL cell lines were treated with pterostilbene (PTE), a non-toxic natural phenolic compound primarily found in blueberries. The antitumor activity of PTE was examined by using the Cell Counting Kit-8, apoptosis assays, cell cycle analysis, JC-1 mitochondrial membrane potential assay, western blot analysis, and tumor xenograft models. PTE treatment induced a dose-dependent inhibition of cell proliferation, including the induction of cell apoptosis and cell cycle arrest at the G0/G1 phase. Moreover, the PI3K/Akt/mTOR pathway was downregulated after PTE treatment, which might account for the anti-MCL effects of PTE. Synergistic cytotoxicity was also observed, both in MCL cells and in xenograft mouse models, when PTE was administered in combination with bortezomib (BTZ). The antitumor effects of PTE shown in our study provide an innovative option for MCL patients with poor responses to standardized therapy. It is noteworthy that the treatment combining PTE with BTZ warrants clinical investigation, which may offer an alternative and effective MCL treatment in the future.
PTE inhibited the PI3K/Akt/mTOR pathway, which may be involved in the antiproliferative mechanism of PTE in MCL cells.
98) 116) Kong, Yuanyuan, et al. “Pterostilbene induces apoptosis and cell cycle arrest in diffuse large B-cell lymphoma cells.” Scientific reports 6 (2016): 37417.
99) 117) Chang, Gaomei, et al. “Pterostilbene induces cell apoptosis and cell cycle arrest in T-cell leukemia/lymphoma by suppressing the ERK1/2 pathway.” BioMed research international 2017 (2017).
100) 118) Dong, J., H. Guo, and Y. Chen. “Pterostilbene induces apoptosis through caspase activation in ovarian cancer cells.” Eur J Gynaecol Oncol 37.3 (2016): 342-7.
101) 119) Chen, Gege, et al. “The blueberry component pterostilbene has potent anti-myeloma activity in bortezomib-resistant cells.” Oncology reports 38.1 (2017): 488-496.
102) Guo, Liying, et al. “Pterostilbene inhibits hepatocellular carcinoma through p53/SOD2/ROS-mediated mitochondrial apoptosis.” Oncology reports 36.6 (2016): 3233-3240.
103) 120) Bin, Wu Hong, et al. “Pterostilbene (3’, 5’-dimethoxy-resveratrol) exerts potent antitumor effects in HeLa human cervical cancer cells via disruption of mitochondrial membrane potential, apoptosis induction and targeting m-TOR/PI3K/Akt signalling pathway.” Journal of BU ON.: official journal of the Balkan Union of Oncology 23.5 (2018): 1384-1389.
104) 121) La Spinaa, Martina, et al. “Pterostilbene Improves Cognitive Performance in Aged Rats: An in Vivo Study.” Cell Physiol Biochem 52 (2019): 232-239.
105) 122) Tan, Kok‑Tong, et al. “Pterostilbene inhibits lung squamous cell carcinoma growth in vitro and in vivo by inducing S phase arrest and apoptosis.” Oncology letters 18.2 (2019): 1631-1640.
Itraconazole
106) 123) Pantziarka, Pan, et al. “Repurposing Drugs in Oncology (ReDO)—itraconazole as an anti-cancer agent.” ecancermedicalscience 9 (2015). free pdf
107) 124) Gu, Juan J., et al. “Itraconazole, an Oral Antifungal Drug, Is Active in Chemotherapy Resistant B-Cell Non-Hodgkin Lymphoma and Enhances the Anti-Tumor Activity of Chemotherapy Agents.” Blood. (2016): 5138-5138.
108) 125) Tsubamoto, Hiroshi, et al. “Repurposing itraconazole as an anticancer agent.” Oncology Letters 14.2 (2017): 1240-1246.
109) 126) Head, Sarah A., et al. “Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells.” Proceedings of the National Academy of Sciences 112.52 (2015): E7276-E7285. Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells.
110) 127) Head, Sarah A., et al. “Simultaneous Targeting of NPC1 and VDAC1 by Itraconazole Leads to Synergistic Inhibition of mTOR Signaling and Angiogenesis.” ACS chemical biology 12.1 (2016): 174-182. Simultaneous targeting of NPC1 and VDAC1 by itraconazole leads to synergistic inhibition of MTOR signaling and angiogenesis
111) 128) Anticancer Res. 2017 Jul;37(7):3521-3526. Itraconazole Modulates Hedgehog, WNT/ß-catenin, as well as Akt Signalling, and Inhibits Proliferation of Cervical Cancer Cells. Ueda T1, Tsubamoto H2,3, Inoue K1, Sakata K1, Shibahara H1, Sonoda T3.
112) 129) Pounds R, Leonard S, Dawson C, Kehoe S. Repurposing itraconazole for the treatment of cancer. Oncology Letters. 2017;14(3):2587-2597.
Metformin DCA Synergy – More references
113) 130) Vial, Guillaume, Dominique Detaille, and Bruno Guigas. “Role of Mitochondria in the Mechanism (s) of Action of Metformin.” Frontiers in endocrinology 10 (2019): 294. Inhibition of Complex 1 by Metformin and Metabolic Reprogramming in Cancer Cells
114) 131) Voltan, Rebecca, et al. “Metformin combined with sodium dichloroacetate promotes B leukemic cell death by suppressing anti-apoptotic protein Mcl-1.” Oncotarget 7.14 (2016): 18965.
115) 132) Li, Bo, et al. “Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells.” Oncotarget 7.37 (2016): 59458.
116) 132) Choi, Yong Won, and In Kyoung Lim. “Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells.” Cancer letters 346.2 (2014): 300-308.
To investigate sensitization of metformin-cytotoxicity, cancer cells were treated with dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase (PDK). Metformin-cytotoxicity was mainly dependent on glucose availability and reducing power generated by pentose phosphate pathway, whereas DCA cotreatment enhanced metformin-cytotoxicity via reprogramming glucose metabolism by inhibiting PDK and increasing mitochondrial respiration. DCA cotreatment elicited cell death rather than cell survival despite high glucose and high GSH condition. In conclusion, DCA sensitized metformin-cytotoxicity by reprogramming glucose metabolism in part from aerobic glycolysis to mitochondrial oxidation, evidenced by measurements of glucose consumption, lactate release, and the ratio of oxygen consumption rate/extracellular acidification rate.
117) 133) Haugrud, Allison B., et al. “Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells.” Breast cancer research and treatment 147.3 (2014): 539-550.
Excellent Review of DCA 2019 !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
118) Tataranni, Tiziana, and Claudia Piccoli. “Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications.” Oxidative Medicine and Cellular Longevity 2019 (2019).Dichloroacetate DCA and Cancer Overview Clinical Applications Tataranni Tiziana Oxidative Med Cel Longevity 2019
DCA PolyNeuropathy
2019 DCA causes neuropathy in NHL case
119) 136) Djordjević, Mladen, and Ivan Petković. “DICHLOROACETATE-INDUCED NEUROPATHY IN HIGH GRADE FOLLICULAR LYMPHOMA PATIENT.” Acta Medica Medianae 58.1 (2019): 102-105. DICHLOROACETATE-INDUCED NEUROPATHY IN HIGH GRADE FOLLICULAR LYMPHOMA PATIENT Djordjević Acta Medica Medianae 2019
DCA Defeats Standard Chemo Drugs
120) 136) Heshe, Dirk, et al. “Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs.” Cancer chemotherapy and pharmacology 67.3 (2011): 647-655.Dichloroacetate defeats cytotoxicity of standard anticancer drugs Heshe Dirk Cancer chemo pharm 2011
121) DCA Poly MVA Combo Paul ANderson
DCA is a relatively small molecule which, in the past, was used as treatment for the metabolic disorder lactic acidosis. It inhibits lactate formation, and via a complex mechanism, switches cell metabolism away from lactic acid production (glycolytic) to mitochondrial energy metabolism (electron transport chain (ETC)).152 Noncancerous human cells prefer this aerobic pathway forenergy formation via ETC use, whereas cancerous cells experience the Warburg Effect, where most glucose is converted to lactate regardless of oxygen availability.153 Forcing a cancerous cellinto ETC use thereby increases damaging oxygen formation and oxygen consumption.154 Because of the differences in metabolism between most cancer cells and normal cells andthrough an intricate mechanism, DCA is able to cause cancer cell death via apoptosis and weaken the cancer cell in lowering its ability to proliferate.
DCA also does this only in cancerous cells and not in normal cells.155156157 In earlier uses of DCA, it was often both overdosed and not administered with appropriatesupport factors. For example, DCA is both a consumer of and dependent upon (for metabolism)glutathione. Glutathione is an important detoxification molecule, and lowering its levels couldslow detoxification and cause toxic side effects.
The bottom line is that DCA is a potent agent in an anticancer strategy, but it must be used in very well-defined dosing parameters and alwayswith support for the metabolic stressors it creates. While most DCA side effects are self-limited(such as dizziness and tingling in hands or feet), in earlier studies where DCA was used at highdose without any supportive therapy (for metabolism and detoxification), people did developnerve inflammation. Dr. Anderson has administered over 10,000 doses of DCA to date withproper support therapies. He has not seen any cases of nerve inflammation, and any other sideeffects patients experienced were very short in duration and all resolved fully.158159160Recently, many scientific papers have continued to explore the mechanisms and potentials of DCA in cancer therapies.161162163164165 Multiple human scientific studies, including drug trialsand case reports,166167168169170171172173174175 have been published with regard to DCA andseveral types of cancer. Currently, DCA is being tested with standard chemotherapy drugs as asynergistic agent.176
It is our belief that DCA (with proper administration and supportive therapies) is an incredible tool in the battle against cancer, and truly is unique in its actions andefficacy.
THE COMBINATION OF POLY-MVA AND DCA
An excellent example of true therapeutic synergy was discovered in the earlier days of Dr.Anderson’s IV research with the combination of Poly-MVA and DCA for both IV and oral use.We will discuss the next level of this therapy with regard to the global metabolic therapyapproach in cancer (Chapter 9). For the IV section, we will describe the basis of the synergy andthe initial case series that was compiled.
Due to the established side effect issues of DCA and considering its mechanism, Dr. Anderson and Dr. Gurdev Parmar postulated two synergistic effects could occur if Poly-MVA and DCAwere used together: mutual anticancer benefit and improvement in the safety and tolerance of DCA. The first step was to have a cell line study done (where cancer cells in a petri dish aretested with each compound individually and then together) to see if the synergy seen “on paper”translated to cancer cell death. The short story is that both Poly-MVA and DCA had tumor kill,but together they had additive benefit, with less DCA being used with the same tumor kill.177
Due to these findings and from prior experience with DCA and Poly-MVA, Dr. Anderson and the IV team at the research center knew how to administer both agents safely so that we couldprovide the therapy without any risk other than those common to other IV therapies. We selecteda group of people with advanced cancer who had failed all therapies (standard oncologytherapies and natural therapies) and consented to this as a trial of unknown outcome (which isoften referred to in oncology research as “salvage therapy”). Over the course of two years, weimplemented the therapy. The original case series is summarized in the table below and was partof the original study, but has not been published separately since reported at the Society ofIntegrative Oncology,178 as is the norm for many studies.
The goal of the DCA and Poly-MVA therapy is to attack the cancer cell where it is weakestvia its unique (but impaired) metabolism relative to normal human cells.179180181182183184185We have used this therapy, and newer versions of it, many times in the years since this study andhave had similar results. In some cases, the protocol involves dietary changes, a small group of oral supplements, and hyperbaric oxygen therapy. Dr. Anderson has never seen these results when using DCA alone. Additionally the rate of side effects from the DCA was drastically reduced such that as of now, nobody has had to dropout of the therapy due to DCA-related side effects. The combined use of DCA and Poly-MVA has been one of the truly big advances in integrative cancer therapies in the past 20 years
122) 31) free pdf 2019 DCA for ALS !!!!
Neurotherapeutics. 2019 Jan;16(1):203-215.Mitochondrial Modulation by Dichloroacetate Reduces Toxicity of Aberrant Glial Cells and Gliosis in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis. Martínez-Palma L1,2, Miquel E3,4, Lagos-Rodríguez V3,4, Barbeito L5, Cassina A4,6, Cassina P7,8. Departamento de Histología y Embriología, Facultad de Medicina, Universidad de la República, Av. Gral Flores 2125, 11800, Montevideo, Uruguay.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by motor neuron (MN) degeneration and gliosis. Neonatal astrocytes obtained from the SOD1G93A rat model of ALS exhibit mitochondrial dysfunction and neurotoxicity that can be reduced by dichloroacetate (DCA), a metabolic modulator that has been used in humans, and shows beneficial effects on disease outcome in SOD1G93A mice. Aberrant glial cells (AbGC) isolated from the spinal cords of adult paralytic SOD1G93A rats exhibit highly proliferative and neurotoxic properties and may contribute to disease progression. Here we analyze the mitochondrial activity of AbGC and whether metabolic modulation would modify their phenotypic profile. Our studies revealed fragmented mitochondria and lower respiratory control ratio in AbGC compared to neonatal SOD1G93A and nontransgenic rat astrocytes. DCA (5 mM) exposure improved AbGC mitochondrial function, reduced their proliferative rate, and importantly, decreased their toxicity to MNs. Furthermore, oral DCA administration (100 mg/kg, 10 days) to symptomatic SOD1G93A rats reduced MN degeneration, gliosis, and the number of GFAP/S100β double-labeled hypertrophic glial cells in the spinal cord. DCA treatment of AbGC reduced extracellular lactate levels indicating that the main recognized DCA action, targeting the pyruvate dehydrogenase kinase/pyruvate dehydrogenase complex, may underlie our findings. Our results show that AbGC metabolic phenotype is related to their toxicity to MNs and indicate that its modulation can reduce glial mediated pathology in the spinal cord. Together with previous findings, these results further support glial metabolic modulation as a valid therapeutic strategy in ALS.
ANtibiotics – Atovaquone, Doxycycline and Clarithromycin inhibit ETC
123) Woodhead, Jeffrey L., et al. “Analyzing the Mechanisms Behind Macrolide Antibiotic-Induced Liver Injury Using Quantitative Systems Toxicology Modeling.” Pharmaceutical research 36.3 (2019): 48.
clarithromycin toxicity is primarily due to inhibition of the mitochondrial electron transport chain (ETC)
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
124) Fiorillo, Marco, et al. “Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells.” Oncotarget 7.23 (2016): 34084.
Atovaquone is an FDA-approved anti-parasitic drug that was first developed to target chloroquine-resistant malaria
Atovaquone can be administered alone as a liquid suspension (brand name Mepron)
Atovaquone is a highly lipophilic compound, with limited solubility in water. The bioavailability of atovaquone is dependent on its formulation and the diet, and its absorption is enhanced by high-fat food intake. Importantly, with current oral formulations, the average serum concentration of atovaquone in humans is > 50 μM.
When atovaquone suspension was administered in humans with food at the standard regimen of 750 mg twice daily, the average steady-state plasma concentration was 21.0 ± 4.9 μg/mL, and the minimum plasma concentration was 16 ± 3.8 μg/mL [29]. It should be noted that in our experiments we have effectively ablated mammosphere formation with 10 μM atovaquone, which corresponds to 3.66 μg/mL. Thus, the clinically relevant, therapeutic plasma concentration of atovaquone is 5-times higher than the concentration that completely blocks the expansion of CSCs.
Atovaquone is an FDA-approved anti-malarial drug, which first became clinically available in the year 2000. Currently, its main usage is for the treatment of pneumocystis pneumonia (PCP) and/or toxoplasmosis in immune-compromised patients. Atovaquone is a hydroxy-1,4-naphthoquinone analogue of ubiquinone, also known as Co-enzyme Q10 (CoQ10). It is a well-tolerated drug that does not cause myelo-suppression. Mechanistically, it is thought to act as a potent and selective OXPHOS inhibitor, by targeting the CoQ10-dependence of mitochondrial complex III. Here, we show for the first time that atovaquone also has anti-cancer activity, directed against Cancer Stem-like Cells (CSCs). More specifically, we demonstrate that atovaquone treatment of MCF7 breast cancer cells inhibits oxygen-consumption and metabolically induces aerobic glycolysis (the Warburg effect), as well as oxidative stress. Remarkably, atovaquone potently inhibits the propagation of MCF7-derived CSCs, with an IC-50 of 1 μM, as measured using the mammosphere assay. Atovaquone also maintains this selectivity and potency in mixed populations of CSCs and non-CSCs. Importantly, these results indicate that glycolysis itself is not sufficient to maintain the proliferation of CSCs, which is instead strictly dependent on mitochondrial function. In addition to targeting the proliferation of CSCs, atovaquone also induces apoptosis in both CD44+/CD24low/− CSC and ALDH+ CSC populations, during exposure to anchorage-independent conditions for 12 hours. However, it has no effect on oxygen consumption in normal human fibroblasts and, in this cellular context, behaves as an anti-inflammatory, consistent with the fact that it is well-tolerated in patients treated for infections. Future studies in xenograft models and human clinical trials may be warranted, as the IC-50 of atovaquone’s action on CSCs (1 μM) is >50 times less than its average serum concentration in humans.
125) Sotgia, Federica, et al. “A mitochondrial based oncology platform for targeting cancer stem cells (CSCs): MITO-ONC-RX.” Cell Cycle 17.17 (2018): 2091-2100.
Repurposing FDA-approved antibiotics for targeting mitochondria in CSCs.
a) Doxycycline;
b) Azithromycin;
c) Pyrvinium (pamoate salt; not shown);
d) Atovaquone; and
e) Bedaquiline. Doxycycline and Azithromycin (a,b) are known to inhibit mitochondrial protein translation as an off-target side effect. They are used clinically as antibiotics to inhibit bacterial protein synthesis. Similarly, Pyrvinium pamoate and Atovaquone (c,d) are known to inhibit OXPHOS (related to mitochondrial complex II/III), as a side effect. Bedaquiline (e) was originally designed to inhibit the bacterial ATP-synthase, which is analogous to mitochondrial complex V. All of these FDA-approved drugs (a-e) have been shown to inhibit the anchorage-independent propagation of CSCs, by targeting mitochondrial function.
126) Srivastava, Indresh K., Hagai Rottenberg, and Akhil B. Vaidya. “Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite.” Journal of Biological Chemistry 272.7 (1997): 3961-3966.
atovaquone, shown to inhibit mitochondrial electron transport,
127) Woodhead, Jeffrey L., et al. “Analyzing the Mechanisms Behind Macrolide Antibiotic-Induced Liver Injury Using Quantitative Systems Toxicology Modeling.” Pharmaceutical research 36.3 (2019): 48.
“clarithromycin toxicity is primarily due to inhibition of the mitochondrial electron transport chain (ETC)”
128) Tan, Qian, et al. “Induction of mitochondrial dysfunction and oxidative damage by antibiotic drug doxycycline enhances the responsiveness of glioblastoma to chemotherapy.” Medical science monitor: international medical journal of experimental and clinical research 23 (2017): 4117.
129) Zhao, Yan, et al. “Doxycycline inhibits proliferation and induces apoptosis of both human papillomavirus positive and negative cervical cancer cell lines.” Canadian journal of physiology and pharmacology 94.5 (2016): 526-533.
130) Ahler, Ethan, et al. “Doxycycline alters metabolism and proliferation of human cell lines.” PloS one 8.5 (2013): e64561.
131)deleted
132) Kolesnik, D. L., et al. “Metformin enhances antitumor action of sodium dichloroacetate against glioma C6.” Experimental oncology 41.2 (2019): 123-129.
133) Kolesnik, D. L., et al. “Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma.” Experimental oncology 41.1 (2019): 14-19.
134) Tian, Dan Dan, et al. “GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial.” Pharmacology Research & Perspectives 7.6 (2019): e00526.
135) Liu, Fengfan, et al. “Improving the anticancer activity of platinum (iv) prodrugs using a dual-targeting strategy with a dichloroacetate axial ligand.” RSC advances 9.39 (2019): 22240-22247.
136) Palamarciuc, Oleg, et al. “Investigation of the cytotoxic potential of methyl imidazole-derived thiosemicarbazones and their copper (ii) complexes with dichloroacetate as a co-ligand.” New Journal of Chemistry 43.3 (2019): 1340-1357.
137) Inanc, Seniz, et al. “Metformin And Dichloroacetate Combination Exert A Synergistic Effect On Cell Viability Of Oral Squamous Cell Carcinoma.” ENT Updates 9.2 (2019): 68-73.
138) Verma, Angela, et al. “Combined use of arginase and dichloroacetate exhibits anti‐proliferative effects in triple negative breast cancer cells.” Journal of Pharmacy and Pharmacology 71.3 (2019): 306-315.
139-140 deleted
DCA and Lactate in ME controls Immune Response
141) Ohashi, Toshimitsu, et al. “Dichloroacetate improves immune dysfunction caused by tumor‐secreted lactic acid and increases antitumor immunoreactivity.” International journal of cancer 133.5 (2013): 1107-1118.
The activation of oncogenic signaling pathways induces the reprogramming of glucose metabolism in tumor cells and increases lactic acid secretion into the tumor microenvironment. This is a well-known characteristic of tumor cells, termed the Warburg effect, and is a candidate target for antitumor therapy. Previous reports show that lactic acid secreted by tumor cells is a proinflammatory mediator that activates the IL-23/IL-17 pathway, thereby inducing inflammation, angiogenesis and tissue remodeling. Here, we show that lactic acid, or more specifically the acidification it causes, increases arginase I (ARG1) expression in macrophages to inhibit T-cell proliferation and activation. Accordingly, we hypothesized that counteraction of the immune effects by lactic acid might suppress tumor development. We show that dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinases, targets macrophages to suppress activation of the IL-23/IL-17 pathway and the expression of ARG1 by lactic acid. Furthermore, lactic acid-pretreated macrophages inhibited CD8+ T-cell proliferation, but CD8+ T-cell proliferation was restored when macrophages were pretreated with lactic acid and DCA. DCA treatment decreased ARG1 expression in tumor-infiltrating immune cells and increased the number of IFN-γ-producing CD8+ T cells and NK cells in tumor-bearing mouse spleen. Although DCA treatment alone did not suppress tumor growth, it increased antitumor immunotherapeutic activity of Poly(IC) in both CD8+ T cell- and NK cell-sensitive tumor models. Therefore, DCA acts not only on tumor cells to suppress glycolysis but also on immune cells to improve the immune status modulated by lactic acid and to increase the effectiveness of antitumor immunotherapy.
142) Santos, Nuno, et al. “Lactate as a Regulator of Cancer Inflammation and Immunity.” Immunometabolism 1.2 (2019).
143) Kim, Jaehong. “Regulation of immune cell functions by metabolic reprogramming.” Journal of immunology research 2018 (2018).
Chrysin an d other Bioiflavonoids
Chrysin inhibits HK2
144) Xu, Dong, et al. “Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2.” Journal of Experimental & Clinical Cancer Research 36.1 (2017): 44.
145) Salimi, Ahmad, et al. “Chrysin as an anti-cancer agent exerts selective toxicity by directly inhibiting mitochondrial complex II and V in CLL B-lymphocytes.” Cancer investigation 35.3 (2017): 174-186.
We investigated the effect of chrysin on isolated normal and chronic lymphocytic leukemia (CLL) B-lymphocytes and their isolated mitochondria. We report that a selective and significant increase in cytotoxicity, intracellular reactive oxygen species, mitochondrial membrane potential collapse, ADP/ATP ratio, caspase 3 activation and finally apoptosis in chrysin-treated CLL B- lymphocytes. Also we determined that chrysin selectively inhibits complex II and ATPases in cancerous mitochondria. In this study we proved that the ability of chrysin to promote apoptosis in CLL B-lymphocytes performed by selectively targeting of mitochondria. Our findings may provide a potential therapeutic approach for using chrysin to target mitochondria in CLL B-lymphocytes.
145) Lim, Whasun, et al. “Chrysin attenuates progression of ovarian cancer cells by regulating signaling cascades and mitochondrial dysfunction.” Journal of cellular physiology 233.4 (2018): 3129-3140.
In contrast with normal cell lines and tissue, HK-2 expression was substantially elevated in the majority of tested HCC cell lines and tumor tissue. Owing to the decrease of HK-2 expression, glucose uptake and lactate production in HCC cells were substantially inhibited after exposure to chrysin. After chrysin treatment, HK-2 which combined with VDAC-1 on mitochondria was significantly declined, resulting in the transfer of Bax from cytoplasm to mitochondria and induction of cell apoptosis. Chrysin-mediated cell apoptosis and glycolysis suppression were dramatically impaired in HK-2 exogenous overexpression cells. Tumor growth in HCC xenograft models was significantly restrained after chrysin treatment and significant decrease of HK-2 expression was observed in chrysin-treated tumor tissue.
Conclusion
Through suppressing glycolysis and inducing apoptosis in HCC, chrysin, or its derivative has a promising potential to be a novel therapeutic for HCC management, especially for those patients with high HK-2 expression.
146) Yao, Junliang, Jingtian Liu, and Wensheng Zhao. “By blocking hexokinase-2 phosphorylation, limonin suppresses tumor glycolysis and induces cell apoptosis in hepatocellular carcinoma.” OncoTargets and therapy 11 (2018): 3793.
147) Li, Wei, et al. “Astragalin reduces hexokinase 2 through increasing miR-125b to inhibit the proliferation of hepatocellular carcinoma cells in vitro and in vivo.” Journal of agricultural and food chemistry 65.29 (2017): 5961-5972.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4537043/
Dai, Weiqi, et al. “By reducing hexokinase 2, resveratrol induces apoptosis in HCC cells addicted to aerobic glycolysis and inhibits tumor growth in mice.” Oncotarget 6.15 (2015): 13703.
148) Naoi, Makoto, et al. “Mitochondria in Neuroprotection by Phytochemicals: Bioactive Polyphenols Modulate Mitochondrial Apoptosis System, Function and Structure.” International journal of molecular sciences 20.10 (2019): 2451.
Resveratrol
149) Han, Youngjin, et al. “Resveratrol as a tumor-suppressive nutraceutical modulating tumor microenvironment and malignant behaviors of cancer.” International journal of molecular sciences 20.4 (2019): 925.
150) Tian, Mengyuan, et al. “Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of VDAC1 by Akt-GSK3 β pathway.” European journal of pharmacology 843 (2019): 80-87.
Our previous studies showed that the effect of resveratrol preventing mitochondrial permeability transition pore (mPTP) opening in myocardial ischemia/reperfusion injury was achieved by regulating voltage-dependent anion channel 1 (VDAC1). However, the underlying mechanism remains unclear. Previous studies demonstrated that the activity and function of VDAC1 are highly regulated by post-translational modification. In present study, we investigated whether resveratrol modulates VDAC1 phosphorylation to achieve cardioprotection and explored the signaling pathways involved. Our findings demonstrated that anoxia/reoxygenation (A/R) treatment, an ischemia/reperfusion model in vitro, enhanced VDAC1 phosphorylation in cardiomyocytes. Moreover, we found phosphorylated VDAC1 showed increased affinity to Bax, whereas interaction with hexokinase 2 (HK2) was reduced. Accordingly, the generation of reactive oxygen species increased, the mitochondrial membrane potential collapsed, mPTP opening increased and cytochrome c released into cytoplasm, thereby leading to increased apoptosis. Moreover, our data showed that pretreatment with resveratrol prior to A/R injury inhibited VDAC1 phosphorylation. Dephosphorylated VDAC1 using pretreated resveratrol promoted dissociation with Bax and binding to HK2, which subsequently protected cardiomyocytes against A/R injury. In addition, Akt and its downstream glycogen synthase kinase 3 β (GSK3β) were phosphorylated by the action of resveratrol. Akt inhibitor IV abrogated Akt-GSK3β phosphorylation and thereby abolished the dephosphorylation activity of resveratrol on VDAC1. Moreover, all resveratrol-mediated protective effects on A/R injured cardiomyocytes were abolished by Akt inhibitor IV. Taken together, our data indicated that A/R injury enhanced VDAC1 phosphorylation in cardiomyocytes, whereas pretreatment with resveratrol dephosphorylated VDAC1 through the Akt-GSK3β pathway, thereby protecting cardiomyocytes against A/R injury.
—————————- —————-
How to measure DCA with a scale
https://www.dcaguide.org/dca-dosage-and-usage-instructions-for-human-cancer-treatment
DCA dosage and usage (Quick guide)
for body weight 161 lbs= and dosage of 20mg per kilogram/day
1462 mg per day in divided doses…….or 730 mg twice a day
———————- ——————–
Facebook Group Judy Dowell is administrator. She went into long term mission from Mantle cell Lymphoma with DCA
Mantle Cell Lymphoma Support for Alternative and/or chemo approach.
Comment by Judy Dowell DCA for MAntle Cell Lymphoma April 19, 2014 at 1:27 PM
In 2006 I was diagnosed with stage 4e mantle cell lymphoma, non-hogekins. It was overwhelming to be told I had a prognosis of six months with or without chemo. It’s a long story how I found dca, but I studied medical and science sites about my cancer, down to the molecular level. I was convinced the orphaned drug creisine (dichloroacetate) was my answer. Abbreviated NaDCA, sodium dichloroacetate dissolves in water and is taken orally once a day. For my 150 pound body weight, 1/8 teaspoon in water was all I needed. I took thiamin (vitamin B1, 500 mg. Daily) to help counter effect the slight neuropothy which completely goes away as soon as you discontinue using DCA. It’s that simple. DCA has proven through medic or human trials to be harmless to the healthy cells. It’s fatal to cancer cells! It reverses the warburg effect so that bad cancer cells realize they are not normal and committee suicide, so the cells die off pretty fast. Dr. Akbar Kahn did human trials at medic or cancer center in Toronto, Canada as well as at the university of Alberta in Edmonton. Go to www.thedcasite for old info from 2007. Check out the medic or cancer center for recent up to the minute info. Dr. Humaira Kahn and Akbar Kahn, as well as others have found that DCA does in fact work. DCA is available today. If your doctor will not appease you by looking into this, find a doctor who will. Chemo makes the doctor $13,000. Per dose, so if your getting rchop or bcnu…chemo, they rake in around $50,000. Per patient! Look around that chemo lab at all the faces, then count how much money that lab is raking in. Compare that to the cost of a sure cure that costs about $100.00 per six months. Get the picture? I spent over $100. In gas to go to the doctor in six months, I cured my cancer with DCA and spent $100. I’m not thrifty, I’m alive which is more than they could offer me!
Judy Dowell-Bundschuh shared a link.
Judy Dowell-Bundschuh I went back on dca since the stem cell transplant only gave me a year remission (2009-2010) and then had four months of b&r (that gets me to 2011) then I went back on dca and i didn’t get any worse while taking dca but I had a ct scan twice, about 4 months apart, then the last ct scan results were that i was finally in remission (Dec 2013). I just had a ct scan today and my Dr called to tell me there is no sign of disease. Yay!
https://www.dcalab.com/
DCAlab.com Our Laboratory: All the products we offer are produced by us in our own laboratory. We are based in EU, have strict quality control and provide Sodium Dichloroacetate of the highest quality. We provide free and fast worldwide shipping. For those in need, we included an option to have DCA delivered by your door in one business day.
http://www.thedcasite.com/dca_dosage.html
The DCA Site dosage calculator
=======================================================
Dr. Allan Magaziner
Magaziner Center for Wellness 1907 Greentree Road Cherry Hill NJ 8003 USA
http://polymvasurvivors.com/testimonial_lymphoma.html
——————————-
Meeting:
AAMP Seattle 2019 Advanced Applications in Medical Practice
October 25, 26 and 27, 2019 Integrative and Naturopathic Oncology for the 21st. Century
listed on Amarc Doctors Directory as Poly MVA USer
Dr. Allan Magaziner
Magaziner Center for Wellness 1907 Greentree Road Cherry Hill NJ 8003
Poly-MVA, AMARC Enterprises (www.polymva.com; 866-POLY-MVA (866-765-9682)).
DCA Dichloroacetate Cancer Treatment Group
Eirin OHagan
I just wanted to let everyone know my DCA treatment for non Hodgkin’s lymphoma has been a success. My tumours have halved after 3 months on dca and my spleen is normal size. Will keep you posted. ??
——————————– ———————–
Peter Thorpe Lori, the best way to dispense is to buy DCA in powder,
I get https://pharma-dca.com/…/100-grams-pure-99-sodium…/ and
then buy a capsule maker from Amazon https://www.amazon.co.uk/…/B006OQ4316/ref=sr_1_1_a_it… and some empty “00” capsules and make your own. Keep powder and capsules in fridge, take one a day along with Vitamin B1 and Alpha Lipoic Acid to protect from peripheral neuropathy. DCA should not cause any side effects except perhaps numbness which is why the other supplements. Good luck, I have stage 4 kidney cancer and now into 5th year so I am convinced DCA has helped.
https://www.facebook.com/dcaguide/?hc_location=ufi
Northern Health Products Tel: 347-480-1188
Email: info@northernhealthproducts.com
===============================================
http://www.dcaguide.org/suppliers/dca-lab
DCA-LAB review
Website: www.dcalab.com
Country: Lithuania, United Stated, Canada
Rating: 5.00
DCA-LAB is an online shop operated by a company Curaltus Ltd., located in Lithuania, their products are stored and shipped from warehouses in United States and Canada. All the details about the company and contact data are present in the website. The company manufactures DCA in it’s own laboratory. The Company provides Certificate of Analysis on every batch of their products.
The company sells Sodium Dichloroacetate (DCA) in powder and capsule form. 333mg and 500mg capsule options and a wide variety of package sizes are available. The prices of their products are lowest of all the reviewed online shops.
DCA-LAB offers Free worldwide shipping via regular post. For an additional fee you can choose express shipping via DHL Express. Express shipping option is available to almost any country in the world. Products are shipped from 3 different warehouses in European Union, Canada and United States, this ensures quick delivery and no custom fees. Paypal payments, Credit/Debit card payments and Direct Bank Transfers are accepted. The website is intuitive and easy to use. Checkout process is simple.
During the conducted laboratory tests, DCA-LAB’s product proved to be the best of all the tested samples. Product is the most pure and contains the least amount of toxic substances. The company claims that Sodium Dichloroacetate they sell is >99.9% pure, actually it is even better – 99.98% pure.
In conclusion, DCA-LAB is a Sodium Dichloroacetate supplier we can recommend. Exceptional quality product, great pricing and free shipping.
===============================================
Mantle Cell Lymphoma Support for Alternative and/or chemo approach. Facebook Group Judy Dowell is administrator. She went into long term mission from Mantle cell Lymphoma with DCA
https://www.facebook.com/groups/9603394299/permalink/10155654596364300/
https://therenodispatch.blogspot.com/2012/10/what-is-dca-real-story-behind-this.html?showComment=1397939232783#c140943722525964779
Judy Dowell April 19, 2014 at 1:27 PM
In 2006 I was diagnosed with stage 4e mantle cell lymphoma, non-hogekins. It was overwhelming to be told I had a prognosis of six months with or without chemo. It’s a long story how I found dca, but I studied medical and science sites about my cancer, down to the molecular level. I was convinced the orphaned drug creisine (dichloroacetate) was my answer. Abbreviated NaDCA, sodium dichloroacetate dissolves in water and is taken orally once a day. For my 150 pound body weight, 1/8 teaspoon in water was all I needed. I took thiamin (vitamin B1, 500 mg. Daily) to help counter effect the slight neuropothy which completely goes away as soon as you discontinue using DCA. It’s that simple. DCA has proven through medic or human trials to be harmless to the healthy cells. It’s fatal to cancer cells! It reverses the warburg effect so that bad cancer cells realize they are not normal and committee suicide, so the cells die off pretty fast. Dr. Akbar Kahn did human trials at medicor cancer center in Toronto, Canada as well as at the university of Alberta in Edmonton. Go to www.thedcasite for old info from 2007. Check out the medic or cancer center for recent up to the minute info. Dr. Humaira Kahn and Akbar Kahn, as well as others have found that DCA does in fact work. DCA is available today. If your doctor will not appease you by looking into this, find a doctor who will. Chemo makes the doctor $13,000. Per dose, so if your getting rchop or bcnu…chemo, they rake in around $50,000. Per patient! Look around that chemo lab at all the faces, then count how much money that lab is raking in. Compare that to the cost of a sure cure that costs about $100.00 per six months. Get the picture? I spent over $100. In gas to go to the doctor in six months, I cured my cancer with DCA and spent $100. I’m not thrifty, I’m alive which is more than they could offer me!
(92) Judy Dowell-Bundschuh shared a link.
https://www.facebook.com/groups/9603394299/permalink/10155654596364300/
Judy Dowell-Bundschuh I went back on dca since the stem cell transplant only gave me a year remission (2009-2010) and then had four months of b&r (that gets me to 2011) then I went back on dca and i didn’t get any worse while taking dca but I had a ct scan twice, about 4 months apart, then the last ct scan results were that i was finally in remission (Dec 2013). I just had a ct scan today and my Dr called to tell me there is no sign of disease. Yay!
(93) DCA LAB
Our Laboratory: All the products we offer are produced by us in our own laboratory. We are based in EU, have strict quality control and provide Sodium Dichloroacetate of the highest quality. We provide free and fast worldwide shipping. For those in need, we included an option to have DCA delivered by your door in one business day.
DCALAB.com Detailed Seller Information
Business Name:UAB “Curaltus”
Business Type:Uždaroji akcine bendrove
Trade Register Number:303419202
VAT Number:LT100010179117
Phone number:+37067680790
Business Address:
Piliakalnio st. 7
Vilnius
VIlniaus m.
06229
LT
===================== ===
Last updated on December 1st, 2019 by
The post DCA Dichloroacetate Breakthrough Anticancer Agent appeared first on Jeffrey Dach MD.



Leave a Comment
You must be logged in to post a comment.