




 How Does Cannabis Kill Cancer Cells ? by Jeffrey Dach MD
How Does Cannabis Kill Cancer Cells ? by Jeffrey Dach MD
An Exciting Time for Medicinal Cannabis
This article is part one. For part two click here.
It is an exciting time for medicinal cannabis, now legal in twenty three states as of June 2014, and widely used by ordinary people as a home cancer treatment. There has been considerable success, and one can read the inspiring stories of remission from cancer on internet Blogs and Message Boards.
Our previous articles in this series have discussed the use of medicinal cannabis oil as curative for leukemia and malignant brain tumors. In this article, we will delve into the exact cellular mechanism by which cannabis kills cancer cells while sparing normal cells.
Above images courtesy of Cover of Molecular Biology of the Cell, 4th edition. Alberts B, Johnson A, Lewis J, et al. New York: Garland Science; 2002. Far left panel: Cell necrosis with rupture of cell membrane and explosive release of cell debris. Middle and right panels are examples of apoptosis, a more controlled process with preservation of cell membrane. The apoptoic cells remain relatively intact while internal organelles are digested and recycled.
Letting Cells Kill Themselves
Strictly speaking, we must confess that medicinal cannabis does not actually “kill” anything, rather, it serves as a messenger that triggers cell signalling pathways leading to programmed cell death via autophagy and apoptosis. In other words, the cancer cell kills itself in a form of suicide, Programmed cell death (PCD) is a normal part of the cell cycle, and a mechanism for destroying unwanted or damaged cells such as cancer cells in the body. Molecular programs intrinsic to the cell are activated to cause its own destruction.
Programmed Cell Death (PCD) – What Is It ?
Another name for Programmed Cell Death (PCD) is “Apoptosis”, a Greek word for “falling away”, coined by Dr J.F. Kerr in his groundbreaking 1972 article on apoptosis.(10) As a normal part of our life cycle, 10 million of our own cells are lost daily via apoptosis, and require replacement cells to take their place. (11) In disease states, there may be disruption in the cellular machinery or signaling pathways that control apoptosis. Excessive apoptosis may cause neurodegenerative disease. Not enough apoptosis may cause cancer.(11)
 Above image: Mouse Paw embryo development showing apoptosis (black arrow) in web area allowing digits to form by “sculpting” the tissue. Courtesy of Scientific American.
Above image: Mouse Paw embryo development showing apoptosis (black arrow) in web area allowing digits to form by “sculpting” the tissue. Courtesy of Scientific American.
Nobel Prize 2002 – Awarded for Work on Apoptosis
The 2002 Nobel Prize in Physiology or Medicine was awarded to Sydney Brenner, H. Robert Horvitz, John E. Sulston for their work on Programmed cell Death, also called “Apoptosis”. This gives you an idea of how important Programmed Cell Death is to biology, health and medicine.
Apoptosis vs. Necrosis
Apoptosis is characterized by cell shrinkage and preservation of the cell membrane which keeps cell contents nice and neat. This must be distinguished from cell necrosis, a more brutal form of cell death caused by toxic injury, such as chemotherapy treatment. Necrotic cells typically show cytoplasmic swelling and rupture of the cell membrane, with release of cellular debris into the surrounding tissues which evokes an inflammatory response. A quote from Molecular Biology of the Cell explains the difference(13)
Cells that die as a result of acute injury typically swell and burst. They spill their contents all over their neighbors—a process called cell necrosis—causing a potentially damaging inflammatory response. By contrast, a cell that undergoes apoptosis dies neatly, without damaging its neighbors. The cell shrinks and condenses. The cytoskeleton collapses, the nuclear envelope disassembles, and the nuclear DNA breaks up into fragments. Most importantly, the cell surface is altered, displaying properties that cause the dying cell to be rapidly phagocytosed, either by a neighboring cell or by a macrophage, before any leakage of its contents occurs. This not only avoids the damaging consequences of cell necrosis but also allows the organic components of the dead cell to be recycled by the cell that ingests it.(13)
Click here for Link to a nice slide show explaining Apoptosis.
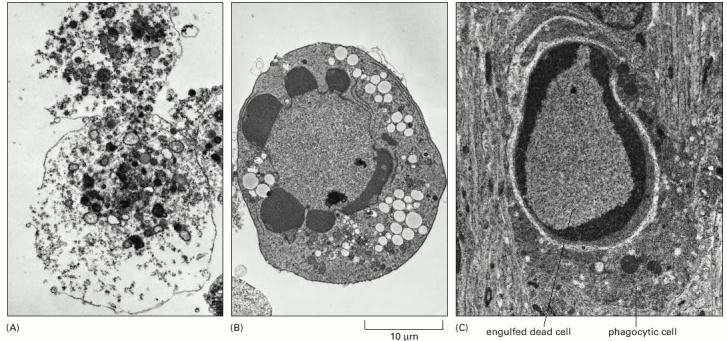
Far left image (A) is a cell that has died from necrosis such as seen from cytotoxic effect of chemotherapy or chemical poison. Notice the cell has “exploded” with debris and cell contents released into surrounding tissues and circulation. Right Images B and C show cells dying from apoptosis, a more controlled process in which there is no release of debris or cell cell contents into surroundings. Dying cells are scavenged by Macrophages. Apoptotic cells in panels (B) and (C) remain relatively intact while undergoing controlled cell death.
Above images from Programmed Cell Death (Apoptosis), courtesy of Cover of Molecular Biology of the Cell, 4th edition. Alberts B, Johnson A, Lewis J, et al. New York: Garland Science; 2002.
The Biology of Cancer
Having the Right Tools
The 2003 Human Genome Project sequenced the entire human genome, a scientific milestone on par with Neal Armstrong’s first step on the moon. Along the way, new research tools of molecular biology were developed. These new molecular biology tools allow us to peer into the inner working of cell biology and glimpse the secrets of the cancer cell.
What Causes Cancer?
If you ask your local doctor the question, “what causes cancer?”, he may tell you “we just don’t know”. This is not entirely correct. We do know. Even the American Cancer society admits that carcinogenic chemicals in the environment cause cancer. If you walk through the hallowed halls of your local university medical school, and ask a cancer researcher, “how do you give your mice cancer? A typical reply: They give the mice carcinogenic chemicals to give the animal cancer. These chemicals cause oxidative damage to the DNA. Of course, our cells are prepared for this and will quickly repair the oxidative damage. However, our repair mechanisms can be overwhelmed, leading to transformation of the normal cell to a cancer cell.
What is a Cancer Cell ? What Makes a Cancer Cell, Cancer ?
Normal cells have a well defined life cycle, starting out as a primitive stem cell, they differentiate into their predestined specialized cell type (liver cell, muscle cell, nerve cell, light receptor cell, red blood cell, etc.) Once differentiated, these mature cells have a limited life span, replicating about fifty times, called the Hayflick Limit. During the life span of a cell, there is an aging process in which cell damage accumulates and causes loss of function. This aging process is caused by oxidative damage to cell organelles, a normal side effect of mitochondrial energy production. Once the cell becomes dysfunctional, internal mechanisms signal it is time for programmed cell death by apoptosis and autophagy, two cell mechanisms we will discuss in more detail below. This end of life event for the cell is called “cell senescence”.
The Hayflick Limit Does Not apply to Cancer Cells
In cell culture, normal cells replicate by means of mitosis about 50 or 60 times, and then are instructed to undergo PCD (programmed cell death). This is called the Hayflick Limit. For cancer cells, the Hayflick Limit does not apply, as cancer cells are immortal and will replicate indefinitely. Cancer cells have escaped the final instruction of programmed cell death. The cancer cell laughs at “cell senescence”, proliferates uncontrollably and is seemingly immune to normal control. This is where the endocannabinoid system and phyto-cannabinoids come in to the picture. Medicinal Cannabis restores the cancer cell’s ability to undergo programmed cell death, which is then automatically triggered, while leaving normal cells unharmed.
Polyploidy and Aneuploidy in Cancer Cells
Normal human cells have 46 chromosomes, while cancer cells have more. This peculiar increased number of chromosomes in cancer cells is called “polyploidy“, and aneuploidy. The number of chromosomes are increased caused by a defect or flaw in the machinery of cell division. The increased nuclear material (chromatin) may be visible on histologic examination with light or electron microscopy. Cancer cells are “genetically unstable” with many other genetic alterations, (chromosomal translocations inversions, gene deletions, gene amplifications, point mutations, duplications and loss of whole chromosomes.) The cancer cell also has alterations in cell size, shape and morphology identified when compared to normal cells in the surrounding tissues.
Other features of cancer cells.
Cancer cells may produce their own growth factors as well as receptors for these growth factors, creating a powerful feedback loop for uncontrolled cell growth and replication. Cancer cells may produce proteolytic enzymes (such as matrix metalloproteases, MMPs) which dissolve the extracellular tissue matrix and allow cancer cells to invade surrounding tissues, blood vessels and lymphatic which are the gateways to metastatic spread. This is described in my previous article on the Trophoblastic theory of Cancer.
See this 2010 article by Mosieniak and Sikora: Polyploidy_Link_Senescence_Cancer_Mosieniak_Sikora_2010
Although the exact mechanism of medicinal cannabis in cancer has remained elusive, recent studies in the laboratories of Drs. Sean McAllister, Shrivastava and Guzman, are closing in on how THC and CBD trigger programmed cell death in the cancer cell.
 Learning the Correct “Ideology”
Learning the Correct “Ideology”
The ID-1 Gene
Sean McAllister and his colleagues at the California Pacific Medical Center in San Francisco report CBD is a novel inhibitor of the ID-1, gene, a key regulator of cellular growth and cell cycle machinery involved in cell differentiation, maturation, senescence and finally programmed cell death.(1-3,19) Dr Sean McAllister’s group also report the combined use of THC with CBD has a synergistic effect with more profound suppression of cancer activity than the use of CBD alone.(4) Above left image courtesy of Sean McAllister.
ID-1 and Cancer Cell Biology
Increased ID-1 expression is associated with a more proliferative and aggressive cancer cell type.(2) In addition, aggressive cancer cell types reverted to normal when the ID-1 gene was targeted and inhibited with anti-sense drug therapy.(2) Sean McAllister’s group discovered that CBD from cannabis down-regulates the ID-1 gene in aggressive cancers, rendering them docile.
ERK-MAPK SIgnalling
The discovery of the ERK/MAPK signaling cascade was a major breakthrough in our understanding of cancer biology and has stimulated intensive efforts by the research community and pharmaceutical industry to develop inhibitors of ERK/MAPK signaling for cancer treatment.(link) (31) Figure courtesy of Wong from AntiCancer_Drugs_Against_Ras_Raf_MEK_ERK_Pathway_2013
Figure courtesy of Wong from AntiCancer_Drugs_Against_Ras_Raf_MEK_ERK_Pathway_2013
The ERK pathway receives signals with receptors located on the outer cell membrane (Yellow). These signals are then transferred to the cell nucleus (Red) ato regulate gene expression. The ERK signalling cascade is involved with cell replication, growth, differentiation, and cell survival. Dysregulation of the ERK pathway leads to dysregulation of ID-1 and this is a common occurrence in human cancers. Sean McAllister’s group reports that CBD up-regulates ERK signaling which inhibits cancer cell proliferation and invasion via down-regulation of ID-1 genetic expression.(1-3)
Mouse Models of Metastatic Cancer
In two mouse models of metastatic cancer treatment with CBD significantly reduces primary tumor mass, size and number of secondary lung metastases .(2)
Dr Sean McAllister says (2):
“The expression of Id-1 protein has been reported to be dysregulated in over 20 types of cancer, and suggested as a key determinant of tumorigenesis and/or metastasis in a wide range of tissues, including the breast [34, 35]. Reducing Id-1 expression (a gene whose expression is absent in most of the healthy adult tissues) could therefore provide a rational therapeutic strategy for the treatment of aggressive cancers.”(2)
 Shrivastava’s Group at Harvard
Shrivastava’s Group at Harvard
Dr Shivastava’s group at Harvard explored the molecular mechanisms by which CBD induced programmed cell death (PCD) in cancer cells. They examined CBD-induced apoptosis, autophagy, and generation of ROS (Reactive Oxygen Species) in multiple human breast cancer cell lines .
Left Image: Courtesy of Jerome Groopman MD, Chair of experimental medicine and chief of Shrivastava’s research group at Beth Israel Hospital..
They found that CBD induced cell death was concentration dependent, and applied equally to (ER+) estrogen receptor-positive and as well as (ER-) breast cancer cell lines. Furthermore, the CBD (cannabidiol) preferentially killed cancer cells, while having no deleterious effect on normal breast cells (MCF-10A cells). The mechanism of action of CBD was independent of cannabinoid receptors CB1, CB2 and Vanniloid receptors. They suggested that the receptor mediating CBD in programmed cell death is yet to be discovered. Their further studies showed that PCD was associated with mitochondrial-mediated apoptosis through both intrinsic and extrinsic cell signalling pathways. Their model also demonstrated that within 2 hours of incubation of the breast cancer cells with CBD, there were signs of ER (endoplasmic reticulum) stress which leads to cell autophagy, in this case, a preclude to programmed cell death.
Autophagy, What is It ?
 Shrivastava’s group found that CBD treated breast cancer cells showed evidence of autophagy, a term meaning “the cell literally eats itself”. “Phage” is derived from the Greek word “phagos”, “to eat”. The characteristic morphologic feature of autophagy shown by examining these cells with the light or electron microscope is the “autophagosome”, a unique double membrane structure in which cell organelles are sequestered and digested, or “eaten”. See schematic image at upper left showing autophagosome double membrane.(6)
Shrivastava’s group found that CBD treated breast cancer cells showed evidence of autophagy, a term meaning “the cell literally eats itself”. “Phage” is derived from the Greek word “phagos”, “to eat”. The characteristic morphologic feature of autophagy shown by examining these cells with the light or electron microscope is the “autophagosome”, a unique double membrane structure in which cell organelles are sequestered and digested, or “eaten”. See schematic image at upper left showing autophagosome double membrane.(6)

Above image shows autophagosome, double membrane vesicle which then fuses with lysosome (which contains oxidative granules) to form the auto-lysosome. Within the auto-lysosome, the organelles undergo fragmentation and degradation.(7)
CBD induces both apoptosis and autophagy

Figure 2. Courtesy of Shrivastava (5)
CBD induces both apoptosis and autophagy in MDA-MB-231 breast cancer cells. A, representative electron micrographs of untreated MDA-MB-231 cells (a) and MDA-MB-231 cells treated with 7.5 μmol/L CBD for 16 hours (b, c, d, and e); N, nucleus; for panels a and b, scale bars, 2 μm. For c, d, and e, scale bars, 500 nm. Panel c, left arrow indicates membrane whorls. Right arrow indicates vacuole filled with degrading organelles. Panel d, double-membraned vacuole fusing with lysosome (indicated by double-headed arrow). Arrow heads indicate empty vacuoles. Panel e, asterisk indicates swollen mitochondrion devoid of cristae. B, representative Western blot analysis of PARP cleavage and LC3 lipidation in MDA-MB-231 cells treated with increasing concentrations of CBD for 24 hours. GAPDH used as loading control. C, percentage of autophagy (as measured by percentage of acridine orange–positive cells) and percentage of apoptosis (as measured by percentage of Annexin V–positive cells) in MDA-MB-231 cells treated with increasing concentrations of CBD for 24 hours. Data represent the mean ± SD of at least 3 independent experiments. (**, P ≤ 0.01).
The machinery of Apoptosis Involves the Mitochondria
Mitochondrial Programmed Cell Death
 Above Diagram shows the intrinsic mitochondrial pathway which involves activation of caspase enzymes cascades, the final step in programmed cell death. Upon trigger event such as CBD activation of the VDAC (voltage dependent anion channedl) (17) , the Mitochondrial outer membrane becomes permeable. This is a point-of-no-return in the mitochondrial apoptotic pathway and induces Mitochondrial release of cytochrome C to the cytosol which then initiates cell death in a caspase-dependent manner. In sddition to proteolytic degradation of cell organelles, proteolytic enzymes are released to the nucleus which induce DNA fragmentation.
Above Diagram shows the intrinsic mitochondrial pathway which involves activation of caspase enzymes cascades, the final step in programmed cell death. Upon trigger event such as CBD activation of the VDAC (voltage dependent anion channedl) (17) , the Mitochondrial outer membrane becomes permeable. This is a point-of-no-return in the mitochondrial apoptotic pathway and induces Mitochondrial release of cytochrome C to the cytosol which then initiates cell death in a caspase-dependent manner. In sddition to proteolytic degradation of cell organelles, proteolytic enzymes are released to the nucleus which induce DNA fragmentation.
AIF, apoptosis-inducing factor; Casp-9, caspase-9; CL, cardiolipin: OPA1, optic atrophy type 1; Cyt. c, cytochrome c; EndoG, endonuclease G; IAP, inhibitor of apoptosis; tBid, truncated Bid. From Perier & Vila, Cold Spring Harbor Perspectives in Medicine (2012). image courtesy of Neurodegenerative Disease Research Lab,
This article is part one. For part two click here.
Articles with Related Interest:
Pediatric Cancer and Cannabis Oil
Cannabis Oil for Acute Leukemia
Medical Cannabis for Epilepsy PArt Two
Medical Cannabis for Epilepsy Part One
Cannabis Oil Brain Tumor remission
Medical Marijuana Research Supressed
Jeffrey Dach MD
7450 Griffin Road, Suite 190
Davie, Fl 33314
954-792-4663
http://www.jeffreydachmd.com
http://www.drdach.com
http://www.naturalmedicine101.com
http://www.bioidenticalhormones101.com
Link to this article: http://wp.me/p3gFbV-1Zz
Links and References
CBD kills breast cancer cells
Sean McAllister and his colleagues at the California Pacific Medical Center in San Francisco reports on how CBD kills breast cancer by down-regulating a gene called ID-1, which is implicated in several types of aggressive cancer. Silencing the ID-1 gene is thus is an excellent strategy for a cancer treatment.
“Cannabidiol offers hope of a non-toxic therapy that could treat aggressive forms of cancer without any of the painful side effects of chemotherapy,” says McAllister.
—————————
free full text
Breast Cancer CBD ID -1 2007 McAllister
1) http://www.ncbi.nlm.nih.gov/
Mol Cancer Ther. 2007 Nov;6(11):2921-7.
Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. McAllister SD1, Christian RT, Horowitz MP, Garcia A, Desprez PY.
Invasion and metastasis of aggressive breast cancer cells is the final and fatal step during cancer progression, and is the least understood genetically. Clinically, there are still limited therapeutic interventions for aggressive and metastatic breast cancers available. Clearly, effective and nontoxic therapies are urgently required. Id-1, an inhibitor of basic helix-loop-helix transcription factors, has recently been shown to be a key regulator of the metastatic potential of breast and additional cancers. Using a mouse model,
Here, we report that cannabidiol (CBD), a cannabinoid with a low-toxicity profile, could down-regulate Id-1 expression in aggressive human breast cancer cells. The CBD concentrations effective at inhibiting Id-1 expression correlated with those used to inhibit the proliferative and invasive phenotype of breast cancer cells. CBD was able to inhibit Id-1 expression at the mRNA and protein level in a concentration-dependent fashion. These effects seemed to occur as the result of an inhibition of the Id-1 gene at the promoter level. Importantly, CBD did not inhibit invasiveness in cells that ectopically expressed Id-1. In conclusion, CBD represents the first nontoxic exogenous agent that can significantly decrease Id-1 expression in metastatic breast cancer cells leading to the down-regulation of tumor aggressiveness.
2011 Mcallister ID-1 Breast Cancer
2) http://www.ncbi.nlm.nih.gov/
Breast Cancer Res Treat. 2011 Aug;129(1):37-47. Epub 2010 Sep 22.
Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis.
McAllister SD1, Murase R, Christian RT, Lau D, Zielinski AJ, Allison J, Almanza C, Pakdel A, Lee J, Limbad C, Liu Y, Debs RJ, Moore DH, Desprez PY.
Abstract
Invasion and metastasis of aggressive breast cancer cells are the final and fatal steps during cancer progression. Clinically, there are still limited therapeutic interventions for aggressive and metastatic breast cancers available. Therefore, effective, targeted, and non-toxic therapies are urgently required. Id-1, an inhibitor of basic helix-loop-helix transcription factors, has recently been shown to be a key regulator of the metastatic potential of breast and additional cancers. We previously reported that cannabidiol (CBD), a cannabinoid with a low toxicity profile, down-regulated Id-1 gene expression in aggressive human breast cancer cells in culture. Using cell proliferation and invasion assays, cell flow cytometry to examine cell cycle and the formation of reactive oxygen species, and Western analysis, we determined pathways leading to the down-regulation of Id-1 expression by CBD and consequently to the inhibition of the proliferative and invasive phenotype of human breast cancer cells. Then, using the mouse 4T1 mammary tumor cell line and the ranksum test, two different syngeneic models of tumor metastasis to the lungs were chosen to determine whether treatment with CBD would reduce metastasis in vivo. We show that CBD inhibits human breast cancer cell proliferation and invasion through differential modulation of the extracellular signal-regulated kinase (ERK) and reactive oxygen species (ROS) pathways, and that both pathways lead to down-regulation of Id-1 expression. Moreover, we demonstrate that CBD up-regulates the pro-differentiation factor, Id-2. Using immune competent mice, we then show that treatment with CBD significantly reduces primary tumor mass as well as the size and number of lung metastatic foci in two models of metastasis. Our data demonstrate the efficacy of CBD in pre-clinical models of breast cancer. The results have the potential to lead to the development of novel non-toxic compounds for the treatment of breast cancer metastasis, and the information gained from these experiments broaden our knowledge of both Id-1 and cannabinoid biology as it pertains to cancer progression.
ID Gene
3) http://www.sciencedirect.com/science/article/pii/S1535610803001417
Cancer Cell. 2003 Jun;3(6):525-30.
Id proteins in cell growth and tumorigenesis.
Sikder HA1, Devlin MK, Dunlap S, Ryu B, Alani RM.
Since the gene encoding Id1 was cloned in 1990, Id proteins have been implicated in regulating a variety of cellular processes, including cellular growth, senescence, differentiation, apoptosis, angiogenesis, and neoplastic transformation. The development of knockout and transgenic animal models for many members of the Id gene family has been particularly useful in sorting out the biologic relevance of these genes and their expression during normal development, malignant transformation, and tumor progression. Here we review the current understanding of Id gene function, the biologic consequences of Id gene expression, and the implications for Id gene regulation of cell growth and tumorigenesis.
Cellular differentiation programs are tightly controlled through the coordinated regulation of gene expression. Basic helix-loop-helix (bHLH) transcription factors regulate the differentiation programs of multiple cell lineages (reviewed in Norton, 2000). These proteins share a common sequence motif of a stretch of basic amino acids responsible for site-specific DNA binding adjacent to a helix-loop-helix dimerization domain. The Id family of helix-loop-helix proteins does not possess a basic DNA binding domain and functions as a dominant-negative regulator of basic HLH proteins through the formation of inactive heterodimers with intact bHLH transcription factors (Figure 1). The Id family of proteins (comprised of 4 members designated Id1–Id4) has been demonstrated to bind the ubiquitously expressed bHLH E-proteins or cell lineage-restricted bHLH transcription factors, leading to inhibition of lineage-specific gene expression and differentiation (Norton et al., 1998). Hence, the name Id refers to both inhibition of differentiation and inhibition of DNA binding. Transcriptional inhibition by Id proteins is mediated via inhibition of DNA binding of bHLH or other activator proteins at E boxes (CANNTG), N boxes (CACNAG), or Ets sites (GGAA/T) present in the promoter regions of regulated genes (reviewed in Zebedee and Hara, 2001). Since cellular differentiation programs are frequently altered during the development of neoplastic disease, it is not surprising that Id proteins would play a role in this process. Indeed, a clue to the potential role of Id genes in tumorigenesis came with the observation that, in general, high Id expression levels are found in proliferative, undifferentiated cells—a feature which is characteristic of tumor cells (Israel et al., 1999). Over the past several years, the particular mechanisms underlying the effects of Id genes on cell growth and differentiation have been investigated. Here we review data supporting the critical role of Id gene regulation in the development of normal cellular differentiation programs. We also review mechanisms of Id gene regulation of cellular growth controls and the cell cycle machinery and evaluate the contribution of dysregulated Id gene expression to the process of tumorigenesis.
4) http://mct.aacrjournals.org/content/9/1/180.short
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2806496/
Mol Cancer Ther. Jan 2010; 9(1): 180–189.
Cannabidiol enhances the inhibitory effects of Δ9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Jahan P. Marcu,1,* Rigel T. Christian,1,* Darryl Lau,1 Anne J. Zielinski,1 Maxx P. Horowitz,1 Jasmine Lee,1 Arash Pakdel,1 Juanita Allison,1 Chandani Limbad,1 Dan H. Moore,1,2 Garret L. Yount,1 Pierre-Yves Desprez,1 and Sean D. McAllister1,3
——————————
Breast Cancer Shrivastava
CBD Breast Cancer Apoptosis and Autophagy
5) http://mct.aacrjournals.org/
Cannabidiol Induces Programmed Cell Death in Breast Cancer Cells by Coordinating the Cross-talk between Apoptosis and Autophagy. Mol Cancer Ther. 2011 Jul;10(7):1161-72. by Ashutosh Shrivastava, Paula M. Kuzontkoski, Jerome E. Groopman, and Anil Prasad Authors’ Affiliation: Division of Experimental Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215
MDA-MB-231 breast cancer cells
In addition, we showed that CBD reduces mitochondrial membrane potential, triggers the translocation of BID to the mitochondria, the release of cytochrome c to the cytosol, and, ultimately, the activation of the intrinsic apoptotic pathway in breast cancer cells.
We found that CBD induced both apoptosis and autophagy in breast cancer cells,
These data enhance the desirability of CBD as an anticancer agent, because they suggest that CBD preferentially kills breast cancer cells, while minimizing damage to normal breast tissue.
Figure 2.
CBD induces both apoptosis and autophagy in MDA-MB-231 breast cancer cells. A, representative electron micrographs of untreated MDA-MB-231 cells (a) and MDA-MB-231 cells treated with 7.5 μmol/L CBD for 16 hours (b, c, d, and e); N, nucleus; for panels a and b, scale bars, 2 μm. For c, d, and e, scale bars, 500 nm. Panel c, left arrow indicates membrane whorls. Right arrow indicates vacuole filled with degrading organelles. Panel d, double-membraned vacuole fusing with lysosome (indicated by double-headed arrow). Arrow heads indicate empty vacuoles. Panel e, asterisk indicates swollen mitochondrion devoid of cristae. B, representative Western blot analysis of PARP cleavage and LC3 lipidation in MDA-MB-231 cells treated with increasing concentrations of CBD for 24 hours. GAPDH used as loading control. C, percentage of autophagy (as measured by percentage of acridine orange–positive cells) and percentage of apoptosis (as measured by percentage of Annexin V–positive cells) in MDA-MB-231 cells treated with increasing concentrations of CBD for 24 hours. Data represent the mean ± SD of at least 3 independent experiments. (**, P ≤ 0.01).
______________________________
Autophagy – double layer lysosome
6) http://myweb.sabanciuniv.edu/
Image
Fig. 3. Electron microscopic (TEM) picture of a double membrane autophagic vesicle (arrow) containing cytoplasm and a degenerating mitochondrion.
figure:
7) http://www.umcutrecht.nl/
Molecular mechanism of autophagosome formation in the yeast Saccharomyces cerevisiae. (with Janice Griffith, in collaboration with Hans Geuze)
Autophagy is a degradative process conserved among all eukaryotic cells and is required for the rapid degradation of large portions of the cytoplasm and unnecessary or damaged organelles in the lysosome lumen. It has long been known that this catabolic pathway is essential to generate an internal pool of nutrients that permit cells to survive during prolonged periods of starvation. Recent studies however, have revealed that autophagy actively participates in other cellular processes such as development, cellular differentiation and rearrangement, aging, elimination of aberrant structures and type II programmed cell death, as well as contributing to the cell’s defense against pathogens (both viruses and bacteria) and tumors. Consequently, defects in this protective barrier correlate with a growing list of diseases, including cancer, neurodegenerative disorders such as Huntington’s, Parkinson’s and Alzheimer’s diseases, and cardiomyopathies.
The main morphological feature of autophagy is the sequestration of the cargo targeted for destruction by a large cytosolic double-membrane vesicle called autophagosome that delivers it into the lysosome/vacuole interior for destruction. Despite the identification of many specific components, the molecular mechanism that directs formation of the sequestering vesicles remains largely unknown.
Conceptual model of autophagy.The cargo destined for destruction is enwrapped by a membrane, leading to the formation of a double-membrane vesicle called an autophagosome. Once the sequestration process is completed, the autophagosome fuses with the lysosome/vacuole. During the fusion event, the external lipid bilayer of the autophagosome becomes part of the lysosome/vacuole surface, whereas the internal vesicle—now termed an autophagic body—is liberated into the interior of this organelle where, together with the cargo, it is degraded by resident hydrolases. [Reggiori F. and Klionsky D.J. (2005), Curr.Opin.Cell.Biol., 17, 415-422]
———
Apoptosis
8) http://www.ncbi.nlm.nih.gov/books/NBK26873/
Programmed Cell Death (Apoptosis) Cover of Molecular Biology of the Cell
Molecular Biology of the Cell. 4th edition. Alberts B, Johnson A, Lewis J, et al.
New York: Garland Science; 2002.
role of mitochondria in programmed cell death
9) http://www.ncbi.nlm.nih.gov/pubmed/11406413 (free full text)
Trends Biochem Sci. 2001 Jun;26(6):390-7.
The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Adrain C1, Martin SJ. Molecular Cell Biology Laboratory, Dept of Genetics, The Smurfit Institute, Trinity College, Dublin 2, Ireland.
The caspase family of cysteine proteases have emerged as central regulators of apoptosis. Diverse cellular stresses trigger caspase activation by promoting release of mitochondrial components, including cytochrome c, into the cytoplasm. In turn, cytochrome c promotes the assembly of a caspase-activating complex termed the apoptosome. In this article, the apoptosome and its role in life and death decisions of cells are discussed.
10) http://www.ncbi.nlm.nih.gov/pubmed/16175394
Cancer Chemother Pharmacol. 2006 May;57(5):545-53.
Role of mitochondria as the gardens of cell death.
Kim R1, Emi M, Tanabe K. 1International Radiation Information Center, Research Institute for Radiation Biology and Medicine, Hiroshima University, 1-2-3 Kasumi Minami-ku, Hiroshima, 734-8553, Japan
Mitochondria play a crucial role in regulating cell death, which is mediated by outer membrane permeabilization in response to death triggers such as DNA damage and growth factor deprivation. Mitochondrial membrane permeabilization induces the release of cytochrome c, Smac/DIABLO, and AIF, which are regulated by proapoptotic and antiapoptotic proteins such as Bax/Bak and Bcl-2/xL in caspase-dependent and caspase-independent apoptosis pathways.
Mitochondrial dysfunction is mediated in two ways. The first is by increased calcium in mitochondria derived from endoplasmic reticulum (ER); this calcium increase is regulated by Bcl-2 and Bax through the ER-mitochondria connection and the unfolded protein response in the ER. The second is by the lysosomal enzyme cathepsin, which activates Bid through lysosome-mitochondria cross-signaling. The genomic responses in intracellular organelles after DNA damage are controlled and amplified in the cross-signaling via mitochondria; such signals induce apoptosis, autophagy, and other cell death pathways. This review discusses the recent advancements in understanding the molecular mechanism of mitochondria-mediated cell death.
11) http://www.ncbi.nlm.nih.gov/pubmed/4561027
Br J Cancer. 1972 Aug;26(4):239-57.
Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. by Kerr JF, Wyllie AH, Currie AR.
The term apoptosis is proposed for a hitherto little recognized mechanism of controlled cell deletion, which appears to play a complementary but opposite role to mitosis in the regulation of animal cell populations. Its morphological features suggest that it is an active, inherently programmed phenomenon, and it has been shown that it can be initiated or inhibited by a variety of environmental stimuli, both physiological and pathological.The structural changes take place in two discrete stages. The first comprises nuclear and cytoplasmic condensation and breaking up of the cell into a number of membrane-bound, ultrastructurally well-preserved fragments. In the second stage these apoptotic bodies are shed from epithelial-lined surfaces or are taken up by other cells, where they undergo a series of changes resembling in vitro autolysis within phagosomes, and are rapidly degraded by lysosomal enzymes derived from the ingesting cells.Apoptosis seems to be involved in cell turnover in many healthy adult tissues and is responsible for focal elimination of cells during normal embryonic development. It occurs spontaneously in untreated malignant neoplasms, and participates in at least some types of therapeutically induced tumour regression. It is implicated in both physiological involution and atrophy of various tissues and organs. It can also be triggered by noxious agents, both in the embryo and adult animal.
12) http://europepmc.org/articles/PMC2117903
Toxicol Pathol. 2007; 35(4): 495–516.
Apoptosis: A Review of Programmed Cell Death Susan Elmore
The process of programmed cell death, or apoptosis, is generally characterized by distinct morphological characteristics and energy-dependent biochemical mechanisms. Apoptosis is considered a vital component of various processes including normal cell turnover, proper development and functioning of the immune system, hormone-dependent atrophy, embryonic development and chemical-induced cell death. Inappropriate apoptosis (either too little or too much) is a factor in many human conditions including neurodegenerative diseases, ischemic damage, autoimmune disorders and many types of cancer. The ability to modulate the life or death of a cell is recognized for its immense therapeutic potential. Therefore, research continues to focus on the elucidation and analysis of the cell cycle machinery and signaling pathways that control cell cycle arrest and apoptosis. To that end, the field of apoptosis research has been moving forward at an alarmingly rapid rate. Although many of the key apoptotic proteins have been identified, the molecular mechanisms of action or inaction of these proteins remain to be elucidated. The goal of this review is to provide a general overview of current knowledge on the process of apoptosis including morphology, biochemistry, the role of apoptosis in health and disease, detection methods, as well as a discussion of potential alternative forms of apoptosis.
13) http://www.ncbi.nlm.nih.gov/books/NBK26873/
Cell Biology 4th Edition Molecular Biology of the Cell. 4th edition.
by Alberts B, Johnson A, Lewis J, et al.
New York: Garland Science; 2002
—————————————
VDAC1 Wikipedia
The voltage-dependent ion channel plays a key role in regulating metabolic and energetic flux across the outer mitochondrial membrane.
Additionally, VDAC is an important regulator of Ca2+ transport in and out of the mitochondria. Because Ca2+ is a cofactor for metabolic enzymes such as pyruvate dehydrogenase and isocitrate dehydrogenase, energetic production and homeostasis are both affected by VDAC’s permeability to Ca2+.
VDAC has also been shown to play a role in apoptosis. [16] During apoptosis, increased permeability of VDAC allows for the release of apoptogenic factors such as cytochrome c. Although cyt. c plays an essential role in oxidative phosphorylation within the mitochondrion, in the cytosol it activates proteolytic enzymes called caspases, which play a major role in cell death.
——————————
Mitochondria Memprane
VDAC in Apoptosis- Regulated by BCL-2
14) http://www.ncbi.nlm.nih.gov/
Biochimie. 2002 Feb-Mar;84(2-3):187-93.
The voltage-dependent anion channel: an essential player in apoptosis. Tsujimoto Y1, Shimizu S.
The increase of outer mitochondrial membrane permeability is a central event in apoptotic cell death, since it releases several apoptogenic factors such as cytochrome c into the cytoplasm that activate the downstream destructive processes. The voltage-dependent anion channel (VDAC or mitochondrial porin) plays an essential role in the increase of mitochondrial membrane permeability, and it is regulated by the Bcl-2 family of proteins via direct interaction.
Anti-apoptotic Bcl-2 family members close the VDAC, whereas some (but not all) pro-apoptotic members interact with the VDAC to generate a protein-conducting channel through which cytochrome c can pass. Although the VDAC is directly involved in the apoptotic increase of mitochondrial membrane permeability and is known to be a component of the permeability transition pore complex, its role in the regulation of outer membrane permeability can be separated from the occurrence of permeability transition events, such as mitochondrial swelling followed by rupture of the outer mitochondrial membrane. The VDAC not only interacts with Bcl-2 family members, but also with other proteins, and probably acts as a convergence point for a variety of life-or-death signals.
——————————
BCL-2
Suppresses apoptosis in a variety of cell systems including factor-dependent lymphohematopoietic and neural cells. Regulates cell death by controlling the mitochondrial membrane permeability. Appears to function in a feedback loop system with caspases. Inhibits caspase activity either by preventing the release of cytochrome c from the mitochondria and/or by binding to the apoptosis-activating factor (APAF-1).
——————————
BCL-2
15) http://www.ncbi.nlm.nih.gov/
Biosci Rep. 2002 Feb;22(1):47-58.
Bcl-2 family of proteins: life-or-death switch in mitochondria.
Tsujimoto Y. Osaka University Medical School, Graduate School of Medicine, CREST of Japanese Science and Technology, Suita.
An increase in the permeability of outer mitochondrial membrane is central to apoptotic cell death, and results in the release of several apoptogenic factors such as cytochrome c into the cytoplasm to activate downstream destructive programs. The voltage-dependent anion channel (VDAC or mitochondrial porin) plays an essential role in disrupting the mitochondrial membrane barrier and is regulated directly by members of the Bcl-2 family proteins. Anti-apoptotic Bcl-2 family members interact with and close the VDAC, whereas some, but not all, proapoptotic members interact with VDAC to open protein-conducting pore through which apoptogenic factors pass. Although the VDAC is involved directly in breaking the mitochondrial membrane barrier and is a known component of the permeability transition pore complex, VDAC-dependent increase in outer membrane permeability can be independent of the permeability transition event such as mitochondrial swelling followed by rupture of the outer mitochondrial membrane. VDAC interacts not only with Bcl-2 family members but also with proteins such as gelsolin, an actin regulatory protein, and appears to be a convergence point for a variety of cell survival and cell death signals
——————————-
BCL-2
16) http://www.nature.com/nrm/journal/v15/n1/full/nrm3722.html
Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy . Nature Reviews Molecular Cell Biology 15, 49–63 (2014) 20 December 2013 by Peter E. Czabotar, Guillaume Lessene, Andreas Strasser . Jerry M. Adams
The BCL-2 protein family determines the commitment of cells to apoptosis, an ancient cell suicide programme that is essential for development, tissue homeostasis and immunity. Too little apoptosis can promote cancer and autoimmune diseases; too much apoptosis can augment ischaemic conditions and drive neurodegeneration. We discuss the biochemical, structural and genetic studies that have clarified how the interplay between members of the BCL-2 family on mitochondria sets the apoptotic threshold. These mechanistic insights into the functions of the BCL-2 family are illuminating the physiological control of apoptosis, the pathological consequences of its dysregulation and the promising search for novel cancer therapies that target the BCL-2 family.
—————————————————————
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
“Thus, VDAC1 seems to serve as a novel mitochondrial target for CBD. The inhibition of VDAC1 by CBD may be responsible for the immunosuppressive and anticancer effects of CBD.“
VDAC and Cannabidiol CBD
17) http://www.ncbi.nlm.nih.gov/
Cell Death Dis. 2013 Dec 5;4:e949.
Direct modulation of the outer mitochondrial membrane channel, voltage-dependent anion channel 1 (VDAC1) by cannabidiol: a novel mechanism for cannabinoid-induced cell death. Rimmerman N1, Ben-Hail D, Porat Z, Juknat A, Kozela E, Daniels MP, Connelly PS, Leishman E, Bradshaw HB, Shoshan-Barmatz V, Vogel Z.The Dr. Miriam and Sheldon G Adelson Center for the Biology of Addictive Diseases, Department of Physiology and Pharmacology, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel.
Abstract
Cannabidiol (CBD) is a non-psychoactive plant cannabinoid that inhibits cell proliferation and induces cell death of cancer cells and activated immune cells. It is not an agonist of the classical CB1/CB2 cannabinoid receptors and the mechanism by which it functions is unknown. Here, we studied the effects of CBD on various mitochondrial functions in BV-2 microglial cells. Our findings indicate that CBD treatment leads to a biphasic increase in intracellular calcium levels and to changes in mitochondrial function and morphology leading to cell death. Density gradient fractionation analysis by mass spectrometry and western blotting showed colocalization of CBD with protein markers of mitochondria. Single-channel recordings of the outer-mitochondrial membrane protein, the voltage-dependent anion channel 1 (VDAC1) functioning in cell energy, metabolic homeostasis and apoptosis revealed that CBD markedly decreases channel conductance. Finally, using microscale thermophoresis, we showed a direct interaction between purified fluorescently labeled VDAC1 and CBD. Thus, VDAC1 seems to serve as a novel mitochondrial target for CBD. The inhibition of VDAC1 by CBD may be responsible for the immunosuppressive and anticancer effects of CBD.
———————————————–
18)
http://sensiseeds.com/en/blog/
How Cannabidiol can prevent the growth and metastasis of breast cancer posted by Seshata on October 19th 2012
——————————
Glioblastoma ID1 McAlister 2013 CBD
19) http://cancerres.aacrjournals.
Id-1 Is a Key Transcriptional Regulator of Glioblastoma Aggressiveness and a Novel Therapeutic Target. Cancer Res. 2013 Mar 1;73(5):1559-69.
Liliana Soroceanu, Ryuichi Murase, Chandani Limbad, Eric Singer, Juanita Allison, Isabel Adrados, Rumi Kawamura, Arash Pakdel, Yayoi Fukuyo, Daniel Nguyen, Sabeena Khan, Robert Arauz, Garret L. Yount, Dan H. Moore, Pierre-Yves Desprez, and Sean D. McAllister Authors’ Affiliation: California Pacific Medical Center, Research Institute, San Francisco, California
Pierre-Yves Desprez, California Pacific Medical Center, Research Institute, 475 Brannan Street, Suite 220, San Francisco, CA 94107.
Abstract Glioblastoma is the most common form of primary adult brain tumors. A majority of glioblastomas grow invasively into distant brain tissue, leading to tumor recurrence, which is ultimately incurable. It is, therefore, essential to discover master regulators that control glioblastoma invasiveness and target them therapeutically. We show here that the transcriptional regulator Id-1 plays a critical role in modulating the invasiveness of glioblastoma cell lines and primary glioblastoma cells. Id-1 expression levels positively correlate with glioma cell invasiveness in culture and with histopathologic grades in patient biopsies. Id-1 knockdown dramatically reduces glioblastoma cell invasion that is accompanied by profound morphologic changes and robust reduction in expression levels of “mesenchymal” markers, as well as inhibition of self-renewal potential and downregulation of glioma stem cell markers. Importantly, genetic knockdown of Id-1 leads to a significant increase in survival in an orthotopic model of human glioblastoma. Furthermore, we show that a nontoxic compound, cannabidiol, significantly downregulates Id-1 gene expression and associated glioma cell invasiveness and self-renewal. In addition, cannabidiol significantly inhibits the invasion of glioblastoma cells through an organotypic brain slice and glioma progression in vivo. Our results suggest that Id-1 regulates multiple tumor-promoting pathways in glioblastoma and that drugs targeting Id-1 represent a novel and promising strategy for improving the therapy and outcome of patients with glioblastoma. Cancer Res; 73(5); 1559–69.
—————————————————-
small cell lung ID1 Chen Lung Cancer 2014
20) mhttp://www.ncbi.nlm.nih.gov/
Genes Cancer. 2014 May;5(5-6):212-25.
Increased expression of Id1 and Id3 promotes tumorigenicity by enhancing angiogenesis and suppressing apoptosis in small cell lung cancer. Chen D, Forootan SS, Gosney JR, Forootan FS, Ke Y.
Molecular Pathology Laboratory, Department of Molecular and Clinical Cancer Medicine, Liverpool University, 5/6th Floor, Duncan Building, Daulby Street, Liverpool, L69 3GA, UK.
Constant deregulation of Id1 and Id3 has been implicated in a wide range of carcinomas. However, underlying molecular evidence for the joint role of Id1 and Id3 in the tumorigenicity of small cell lung cancer (SCLC) is sparse. Investigating the biological significance of elevated expression in SCLC cells, we found that Id1 and Id3 co-suppression resulted in significant reduction of proliferation rate, invasiveness and anchorage-independent growth. Suppressing both Id1 and Id3 expression also greatly reduced the average size of tumors produced by transfectant cells when inoculated subcutaneously into nude mice. Further investigation revealed that suppressed expression of Id1 and Id3 was accompanied by decreased angiogenesis and increased apoptosis. Therefore, the SCLC tumorigenicity suppression effect of double knockdown of Id1 and Id3 may be regulated through pathways of apoptosis and angiogenesis.
———————–
Breast Cancer DiMarzo Italy 2006
21) http://jpet.aspetjournals.org/
Antitumor Activity of Plant Cannabinoids with Emphasis on the Effect of Cannabidiol on Human Breast Carcinoma. J Pharmacol Exp Ther. 2006 Sep;318(3):1375-87. Epub 2006 May 25.by Alessia Ligresti,Aniello Schiano Moriello,Katarzyna Starowicz,
Isabel Matias,Simona Pisanti,Luciano De Petrocellis,
Chiara Laezza,Giuseppe Portella,Maurizio Bifulco and Vincenzo Di Marzo -Endocannabinoid Research Group, Dr. Vincenzo Di Marzo, Istituto di Chimica Biomolecolare, Consiglio Nazionale delle Ricerche, Via Campi Flegrei 34, 80078 Pozzuoli, Napoli, Italy.
In a 2006 study, various human breast cancer cells (including the most aggressive known cell line, MDA-MB231) were treated in vitro with five natural phytocannabinoid compounds – cannabidiol, cannabigerol, cannabichromene, cannabidiol acid and THC acid. Cannabidiol was shown to be the most effective inhibitor of neoplastic cell proliferation, and was effective even at very low molecular concentration.
In conclusion, our data indicate that cannabidiol, and possibly Cannabis extracts enriched in this natural cannabinoid, represent a promising nonpsychoactive antineoplastic strategy. In particular, for a highly malignant human breast carcinoma cell line, we have shown here that cannabidiol and a cannabidiol-rich extract counteract cell growth both in vivo and in vitro as well as tumor metastasis in vivo. Cannabidiol exerts its effects on these cells through a combination of mechanisms that include either direct or indirect activation of CB2 and TRPV1 receptors and induction of oxidative stress, all contributing to induce apoptosis.
—————————————————
22) http://sensiseeds.com/en/blog/
Born and raised in England, Seshata moved to Amsterdam in 2004, immediately becoming involved in the cannabis industry.
23) http://sensiseeds.com/en/blog/
How Cannabidiol can prevent the growth and metastasis of breast cancer posted by Seshata on October 19th 2012
=========================
Review 2014 Cannabinoids in Cancer
24) http://www.impactjournals.com/
free full
Oncotarget. 2014 Aug 15;5(15):5852-72.
Cannabinoids as therapeutic agents in cancer: current status and future implications. Chakravarti B1, Ravi J2, Ganju RK3.
The pharmacological importance of cannabinoids has been in study for several years. Cannabinoids comprise of (a) the active compounds of the Cannabis sativa plant, (b) endogenous as well as (c) synthetic cannabinoids. Though cannabinoids are clinicall`y used for anti-palliative effects, recent studies open a promising possibility as anti-cancer agents. They have been shown to possess anti-proliferative and anti-angiogenic effects in vitro as well as in vivo in different cancer models. Cannabinoids regulate key cell signaling pathways that are involved in cell survival, invasion, angiogenesis, metastasis, etc. There is more focus on CB1 and CB2, the two cannabinoid receptors which are activated by most of the cannabinoids. In this review article, we will focus on a broad range of cannabinoids, their receptor dependent and receptor independent functional roles against various cancer types with respect to growth, metastasis, energy metabolism, immune environment, stemness and future perspectives in exploring new possible therapeutic opportunities.
Cancer is caused by uncontrolled proliferation of cells and the ability of these cells to invade into other tissues and spread.
Role of cannabinoids in regulation of cancer growth
One of the important aspects of an effective anti-tumor drug is its ability to inhibit proliferation of cancer cells. Cancer cells proliferate rapidly in uncontrolled manner. Also, these cells escape death mechanism which a normal cell undergoes like apoptosis. Apoptosis is a kind of programmed cell death (PCD) mechanism which involves activation of caspase dependent and independent pathways [39]. Cannabinoids have been proved to be anti-proliferative and apoptotic drugs. This section comprises of the detailed role of cannabinoids in modulation of tumor proliferation, cell cycle and apoptosis in various cancer types.
==============================
ID helix-loop-helix pathway
25) http://www.ncbi.nlm.nih.gov/
Differentiation. 2006 Dec;74(9-10):481-7.
The multiple roles of Id-1 in cancer progression.
Ling MT1, Wang X, Zhang X, Wong YC. 1Cancer Biology Group, Department of Anatomy, The University of Hong Kong, Hong Kong, China.
Id-1 (Inhibitor of differentiation/DNA binding) is a member of the helix-loop-helix protein family expressed in actively proliferating cells. It regulates gene transcription by heterodimerization with the basic helix-loop-helix transcription factors and therefore inhibits them from DNA binding and transactivation of their target genes. Early studies showed that Id-1 functions mainly as a regulator in cellular differentiation of the muscle cells. The oncogenic role of Id-1 was revealed recently by the finding that Id-1 expression was able to induce cancer cell growth and promote cell survival. In addition, Id-1 protein was frequently overexpressed in over 20 types of cancer, supporting its role in the tumorigenesis of a wide range of tissues. However, the fact that Id-1 was able to activate multiple pathways involved in tumor progression suggests that Id-1 may in addition function in promotion of tumor development. For example, overexpression of Id-1 was found to induce expression of MT1-MMP protein, leading to invasion of breast cancer cells. A close association between Id-1 expression and angiogenesis has also been demonstrated recently in both normal and cancer cells. Accordingly, in prostate cancer cells, expression of Id-1 was able to activate EGF-R and nuclear factor-kappaB activities and resulted in progression to androgen independence. In addition, in both nasopharyngeal carcinoma and prostate cancer cells, Id-1 expression was found to protect the cells from chemotherapeutic drug-induced apoptosis through regulation of the Raf-1/MAPK and JNK pathways. This review will discuss recent evidence supporting the role of Id-1 in tumor progression and the mechanisms involved.
26) http://jcs.biologists.org/
J Cell Sci. 2000 Nov;113 ( Pt 22):3897-905.
ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. Norton JD.
The ubiquitously expressed family of ID helix-loop-helix (HLH) proteins function as dominant negative regulators of basic HLH (bHLH) transcriptional regulators that drive cell lineage commitment and differentiation in metazoa. Recent data from cell line and in vivo studies have implicated the functions of ID proteins in other cellular processes besides negative regulation of cell differentiation. ID proteins play key roles in the regulation of lineage commitment, cell fate decisions and in the timing of differentiation during neurogenesis, lymphopoiesis and neovascularisation (angiogenesis). They are essential for embryogenesis and for cell cycle progression, and they function as positive regulators of cell proliferation. ID proteins also possess pro-apoptotic properties in a variety of cell types and function as cooperating or dominant oncoproteins in immortalisation of rodent and human cells and in tumour induction in Id-transgenic mice. In several human tumour types, the expression of ID proteins is deregulated, and loss- and gain-of-function studies implicate ID functions in the regulation of tumour growth, vascularisation, invasiveness and metastasis. More recent biochemical studies have also revealed an emerging ‘molecular promiscuity’ of mammalian ID proteins: they directly interact with and modulate the activities of several other families of transcriptional regulator, besides bHLH proteins.
ID PROTEINS IN CELL CYCLE CONTROL
Genetic studies in Drosophila and mice
(discussed above) now provide direct evidence for a role for
ID proteins in cell proliferation and cell cycle control in vivo.
Quiescent cells express low/undetectable levels of Id genes.
However, following mitogenic stimulation of, for example,
fibroblasts, Id expression is rapidly induced (within 1-2 hours)
ID proteins in primary tumours
Immortalised cell lines from a number of different tumour
types display markedly elevated levels of one or more Id
mRNAs (reviewed in Israel et al., 1999).
Table 3 lists the major human tumour types in which
ID protein levels have been reported to be deregulated.
Table 3. Primary human tumour types in which ID expression is deregulated
Tumour type References
Pancreatic cancer Kleef et al., 1998; Maruyama et al., 1999
Astrocytic tumours Andres-Barquin et al., 1997
High-grade neural tumour Lyden et al., 1999 endothelial cells
Invasive breast carcinoma Lin et al., 2000
Seminomas Sablitzky et al., 1998
Colorectal adenocarcinoma J. Wilson and J. Norton, unpublished observations
Multiple myeloma L. Peterson and J. Norton, unpublishe
As mentioned previously, the Id1+/- Id3-/- knockout mouse,
although viable, has a severely impaired ability to support the
growth and metastasis of tumour xenografts, owing to a defect
in tumour cell vascularisation (Lyden et al., 1999). Significant
regression is observed for some tumour types even in mice
lacking just one Id allele (Id1+/- Id3+/+), which implies that
there is a gene-dosage effect on ability to support tumour
xenografts. The ability to support tumour growth in Id1-Id3-
deficient mice is also paralleled by impairment in the ability to
support metastasis.
!!!!
Inhibition of cell differentiation through antagonism of bHLH proteins, as originally described ten years ago, still remains the single most important facet of ‘Ideology’.
ID proteins regulate the cell cycle machinery.
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
2013 – ID1 knockdown, cannabinoids and Glioblastoma (Sean D. McAllister)
this is a duplicate of reference (17)
27) http://cancerres.aacrjournals.
Cancer Res. 2013 Mar 1;73(5):1559-69.
Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target.Soroceanu L1, Murase R, Limbad C, Singer E, Allison J, Adrados I, Kawamura R, Pakdel A, Fukuyo Y, Nguyen D, Khan S, Arauz R, Yount GL, Moore DH, Desprez PY, McAllister SD.
Glioblastoma is the most common form of primary adult brain tumors. A majority of glioblastomas grow invasively into distant brain tissue, leading to tumor recurrence, which is ultimately incurable. It is, therefore, essential to discover master regulators that control glioblastoma invasiveness and target them therapeutically. We show here that the transcriptional regulator Id-1 plays a critical role in modulating the invasiveness of glioblastoma cell lines and primary glioblastoma cells. Id-1 expression levels positively correlate with glioma cell invasiveness in culture and with histopathologic grades in patient biopsies. Id-1 knockdown dramatically reduces glioblastoma cell invasion that is accompanied by profound morphologic changes and robust reduction in expression levels of “mesenchymal” markers, as well as inhibition of self-renewal potential and downregulation of glioma stem cell markers. Importantly, genetic knockdown of Id-1 leads to a significant increase in survival in an orthotopic model of human glioblastoma. Furthermore,
!!!!!!!!!!
we show that a nontoxic compound, cannabidiol, significantly downregulates Id-1 gene expression and associated glioma cell invasiveness and self-renewal. In addition, cannabidiol significantly inhibits the invasion of glioblastoma cells through an organotypic brain slice and glioma progression in vivo. Our results suggest that Id-1 regulates multiple tumor-promoting pathways in glioblastoma and that drugs targeting Id-1 represent a novel and promising strategy for improving the therapy and outcome of patients with glioblastoma.
Cannabidiol inhibits Id-1 gene expression and corresponding brain cancer cell invasiveness of U251 cells and primary glioblastoma cells
We have recently shown in culture that cannabidiol was an effective inhibitor of Id-1 expression and corresponding breast cancer cell aggressiveness, that is, invasion and proliferation (25, 26). To determine whether cannabidiol could inhibit Id-1 expression in aggressive brain cancers, U251 cells were treated with cannabidiol for 3 days and analyzed for Id-1 protein using Western blot analysis. In U251 cells, cannabidiol produced a concentration-dependent downregulation of Id-1 (Fig. 6A). In addition, the downregulation of Id-1 expression correlated with a concentration-dependent inhibition of U251 cell invasion (Fig. 6B). Similar activity was observed in primary glioblastoma cells that express Id-1 (Fig. 6C and D). Moreover, cannabidiol modulated the phosphorylation of several phospho-kinases in U251 cells, including AKT
Cannabidiol significantly inhibits glioblastoma dispersal ex vivo and reduces tumorigenicity and Id-1 expression in vivo
To determine whether cannabidiol could inhibit glioblastoma cell invasion through intact brain tissue, we used an organotypic brain slice assay (27). GFP-labeled U251 cells were treated with vehicle or cannabidiol and cells that successfully invaded through the slice were visualized using an inverted microscope.
We found that cannabidiol was highly effective at inhibiting invasion of U251 cells
!!!!!!!!!!!!!!!!!!
ZZZZZZZZZZZZZZZZZZZZZZZZZZZZZZ
tumors were generated in athymic nu/nu mice by intracranial injection of U251 glioblastoma cells. Seven days after tumor implantation, mice were injected systemically (intraperitoneal) with 15 mg/kg cannabidiol 5 d/wk for 28 days until vehicle-treated animals showed signs of significant disease progression, when all mice in the study were euthanized to compare tumor growth. Cannabidiol produced a robust reduction of glioblastoma progression, decreasing the tumor area by 95% (Fig. 7B and C).
In addition to the intracranial model, we conducted a longitudinal assessment of the efficacy of cannabidiol in a subcutaneous model of glioblastoma (Supplementary Fig. S5). Again, cannabidiol significantly reduced tumor progression and also inhibited the expression of Id-1 and Ki67. Similar to the intracranial model, cannabidiol eradicated the tumor in 1 of the 5 mice treated. Overall, cannabidiol was highly effective at reducing Id-1 expression and aggressiveness in cancer cells in culture as well as downregulating Id-1 expression and tumorigenesis in vivo.
In U251 cells, Id-1 knockdown led to almost complete inhibition of cell invasion but only a modest reduction in cell growth. Even more striking was the profound change in morphology produced in this cell population where the cells rounded up into grape-like cluster. These changes suggested induction of a cellular differentiated state.
Using a phospho-kinase array, we also found that multiple proteins associated with tumor aggressiveness were downregulated upon knockdown of Id-1, notably p-ERK and p-AKT pathways.
With its lack of systemic toxicity and psychoactivity, cannabidiol is an ideal candidate agent in this regard and may prove useful in combination with front-line agents for the treatment of patients with aggressive and high-grade glioblastoma tumors.
______________________________
Breast Cancer IDF ` Macallister group cannabidiol
28) http://www.ncbi.nlm.nih.gov/
Breast Cancer Res Treat. 2011 Aug;129(1):37-47. doi: 10.1007/s10549-010-1177-4. Epub 2010 Sep 22.
Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis.
McAllister SD1, Murase R, Christian RT, Lau D, Zielinski AJ, Allison J, Almanza C, Pakdel A, Lee J, Limbad C, Liu Y, Debs RJ, Moore DH, Desprez PY.
1California Pacific Medical Center, Research Institute, San Francisco, CA 94107, USA.
Invasion and metastasis of aggressive breast cancer cells are the final and fatal steps during cancer progression. Clinically, there are still limited therapeutic interventions for aggressive and metastatic breast cancers available. Therefore, effective, targeted, and non-toxic therapies are urgently required. Id-1, an inhibitor of basic helix-loop-helix transcription factors, has recently been shown to be a key regulator of the metastatic potential of breast and additional cancers. We previously reported that cannabidiol (CBD), a cannabinoid with a low toxicity profile, down-regulated Id-1 gene expression in aggressive human breast cancer cells in culture. Using cell proliferation and invasion assays, cell flow cytometry to examine cell cycle and the formation of reactive oxygen species, and Western analysis, we determined pathways leading to the down-regulation of Id-1 expression by CBD and consequently to the inhibition of the proliferative and invasive phenotype of human breast cancer cells. Then, using the mouse 4T1 mammary tumor cell line and the ranksum test, two different syngeneic models of tumor metastasis to the lungs were chosen to determine whether treatment with CBD would reduce metastasis in vivo. We show that CBD inhibits human breast cancer cell proliferation and invasion through differential modulation of the extracellular signal-regulated kinase (ERK) and reactive oxygen species (ROS) pathways, and that both pathways lead to down-regulation of Id-1 expression. Moreover, we demonstrate that CBD up-regulates the pro-differentiation factor, Id-2.
Using immune competent mice, we then show that treatment with CBD significantly reduces primary tumor mass as well as the size and number of lung metastatic foci in two models of metastasis. Our data demonstrate the efficacy of CBD in pre-clinical models of breast cancer. The results have the potential to lead to the development of novel non-toxic compounds for the treatment of breast cancer metastasis, and the information gained from these experiments broaden our knowledge of both Id-1 and cannabinoid biology as it pertains to cancer progression.
29) http://mct.aacrjournals.org/
Mol Cancer Ther. 2007 Nov;6(11):2921-7.
Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells.
McAllister SD1, Christian RT, Horowitz MP, Garcia A, Desprez PY.
Invasion and metastasis of aggressive breast cancer cells is the final and fatal step during cancer progression, and is the least understood genetically. Clinically, there are still limited therapeutic interventions for aggressive and metastatic breast cancers available. Clearly, effective and nontoxic therapies are urgently required. Id-1, an inhibitor of basic helix-loop-helix transcription factors, has recently been shown to be a key regulator of the metastatic potential of breast and additional cancers. Using a mouse model, we previously determined that metastatic breast cancer cells became significantly less invasive in vitro and less metastatic in vivo when Id-1 was down-regulated by stable transduction with antisense Id-1. It is not possible at this point, however, to use antisense technology to reduce Id-1 expression in patients with metastatic breast cancer. Here, we report that cannabidiol (CBD), a cannabinoid with a low-toxicity profile, could down-regulate Id-1 expression in aggressive human breast cancer cells. The CBD concentrations effective at inhibiting Id-1 expression correlated with those used to inhibit the proliferative and invasive phenotype of breast cancer cells. CBD was able to inhibit Id-1 expression at the mRNA and protein level in a concentration-dependent fashion. These effects seemed to occur as the result of an inhibition of the Id-1 gene at the promoter level. Importantly, CBD did not inhibit invasiveness in cells that ectopically expressed Id-1. In conclusion, CBD represents the first nontoxic exogenous agent that can significantly decrease Id-1 expression in metastatic breast cancer cells leading to the down-regulation of tumor aggressiveness.
Furthermore, Id-1 overexpression in breast cancer cells was also found to be one of the most significant genes within a gene signature set that is correlated with the propensity of primary human breast cancer cells to metastasize to the lung
We used the human breast cancer cells lines MDA-MB231 and MDA-MB436
All three compounds tested, i.e., CBD, Δ9-THC, and WIN55,212-2, significantly reduced the invasion of MDA-MB231 cells (Fig. 1A ). Again, as was observed with the cell proliferation experiments, the most potent inhibitor of invasion was CBD.
Figure 1. saved image
CBD is the most effective inhibitor of invasiveness and Id-1 expression in MDA-MD231 cells. A, the Boyden chamber invasion assay was used to determine the effects of cannabinoids on the invasiveness of aggressive human breast cancer MDA-MB231 cells. Compounds were added at concentrations of 0.1, 1.0, or 1.5 μmol/L. Data are presented as relative invasiveness of the cells through the Matrigel, where the respective controls are set as 100%. B, proteins from MDA-MB231 cells treated with vehicle (control), 0.1, 1.0, or 1.5 μmol/L of CBD for 3 d were extracted and analyzed for Id-1 by Western blot analysis as described in Materials and Methods. C, proteins from MDA-MB231 cells treated with additional cannabinoids for 3 d were extracted and analyzed for Id-1 by Western blot analysis. Normalization was carried out by stripping the blots and reprobing with a monoclonal antitubulin antibody. Densitometry readings of the blots were taken and the percentage of relative expression was calculated as the expression of Id-1 in the treated cells / vehicle cells × 100. D, the inhibitory effect of 1.5 μmol/L of CBD on Id-1 expression was compared over a time course of 1, 2, and 3 d. Columns, mean of at least three replicates; bars, SE. Data were compared using a one-way ANOVA with a corresponding Dunnett’s post hoc test. *, P < 0.05, statistically significant differences from
CBD Down-regulates Id-1 Expression
We predicted that CBD, the most potent inhibitor of breast cancer cell proliferation and invasion tested, would regulate the expression of key genes that control breast cancer cell proliferation and invasiveness. A potential candidate protein that could mediate the effects of CBD on both phenotypes was the helix-loop-helix protein Id-1. We determined that treatment of MDA-MB231 cells with CBD led to a concentration-dependent inhibition of Id-1 protein expression (Fig. 1B and C). The inhibitory effect of CBD on Id-1 expression occurred at concentrations as low as 100 nmol/L. CBD was significantly more effective at reducing Id-1 protein expression compared with other cannabinoid compounds (Fig. 1C). The CBD concentrations effective at inhibiting Id-1 expression correlated with those used to inhibit the proliferative and invasive phenotype of MDA-MB231 cells. Furthermore, the down-regulation of Id-1 protein in the presence of CBD seemed to precede, and not follow, the inhibitory effects of CBD on the proliferation and invasiveness of MDA-MB231 cells (Fig. 1D), suggesting that Id-1 down-regulation represents a cause rather than a consequence of a decrease in breast cancer cell aggressiveness.
CBD Inhibits the Transcription of the Id-1 Gene
Id-1 mRNA expression was significantly reduced upon treatment with CBD in MDA-MB231 cells.
Metastasis is the final and fatal step in the progression of breast cancer. Currently available therapeutic strategies at this stage of cancer progression are often nonspecific, have only marginal efficacy, and are highly toxic
We propose that the use of CBD, as an inhibitor of Id-1, represents a novel strategy to treat breast cancer. A wide range of cannabinoid compounds were tested and CBD, a nonpsychoactive cannabinoid constituent, was the most potent inhibitor of human breast cancer cell aggressiveness through Id-1 mRNA and protein down-regulation.
Cannabinoid agonists working through CB1 and CB2 receptors have been shown to act as tumor inhibitors in a variety of cancer models (7, 8). Present evidence also shows that the cannabinoid constituent CBD, which has negligible affinity for CB1 and CB2 receptors, also has antitumor activity
As presented in Fig. 3, CBD seems to act by down-regulating endogenous Id-1 gene expression at the promoter level,
These data indicate that Id-1 is a key factor whose expression needs to be down-regulated in order to observe the beneficial effects of CBD on the reduction of breast cancer cell aggressiveness. Based on previous findings (reviewed in ref. 15), we suggest that a decrease in Id-1 protein upon CBD treatment might consequently lead to a down-regulation of growth-promoting genes such as Zfp289 as well as to a down-regulation of invasion-promoting genes such as the membrane type matrix metalloproteinase (MT1-MMP).
30) http://www.ncbi.nlm.nih.gov/
Adv Exp Med Biol. 2013;986:209-20. doi: 10.1007/978-94-007-4719-7_11.
Cannabinoid signaling in glioma cells.
Ellert-Miklaszewska A1, Ciechomska I, Kaminska B.
Cannabinoids are a group of structurally heterogeneous but pharmacologically related compounds, including plant-derived cannabinoids, synthetic substances and endogenous cannabinoids, such as anandamide and 2-arachidonoylglycerol. Cannabinoids elicit a wide range of central and peripheral effects mostly mediated through cannabinoid receptors. There are two types of specific G(i/o)-protein-coupled receptors cloned so far, called CB1 and CB2, although an existence of additional cannabinoid-binding receptors has been suggested. CB1 and CB2 differ in their predicted amino acid sequence, tissue distribution, physiological role and signaling mechanisms. Significant alterations of a balance in the cannabinoid system between the levels of endogenous ligands and their receptors occur during malignant transformation in various types of cancer, including gliomas. Cannabinoids exert anti-proliferative action in tumor cells. Induction of cell death by cannabinoid treatment relies on the generation of a pro-apoptotic sphingolipid ceramide and disruption of signaling pathways crucial for regulation of cellular proliferation, differentiation or apoptosis. Increased ceramide levels lead also to ER-stress and autophagy in drug-treated glioblastoma cells.
====================
The MAPK/ERK pathway
The MAPK/ERK pathway (also known as the Ras-Raf-MEK-ERK pathway) is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell.
MAPK (mitogen-activated protein kinases, originally called ERK, extracellular signal-regulated kinases),
Uncontrolled growth is a necessary step for the development of all cancers.[6] In many cancers (e.g. melanoma), a defect in the MAP/ERK pathway leads to that uncontrolled growth. Many compounds can inhibit steps in the MAP/ERK pathway, and therefore are potential drugs for treating cancer
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
figure 3 showing Diagram of ERK pathways and drugs in development
Full pdf downloaded
31) Anti_Cancer_Drugs_Against_Ras_Raf_MEK_ERK_Pathway_Wong_2013Crane,
The Therapeutic Promise of Anti-Cancer Drugs Against the Ras/Raf/MEK/ERK Pathway Erin K. Crane and Kwong-Kwok Wong*
Department of Gynecologic Oncology and Reproductive Medicine, The University of Texas, M.D. Anderson Cancer Center, Houston, Texas, USA
Abstract: The Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) pathway mediates cellular responses to different growth signals and is frequently deregulated in cancer. There are three Raf kinases-A-Raf, B-Raf, and C-Raf; however, only B-Raf is frequently mutated in various cancers. The most common B-Raf mutation involves a substitution of a glutamic acid residue to a valine moiety at codon 600. Subsequently, the MAPK pathway is constitutively activated, even in the absence of any growth signals. Although early attempts to target Ras have not yielded any viable drug candidates, many novel compounds inhibiting the activities of B-Raf and MEK have been developed and investigated in clinical trials in recent years and have shown promising result. The first MEK inhibitor (CI-1040) lacked efficacy in clinical trials, but its low toxicity encouraged the search for novel compounds-now there are over a hundred open clinical trials employing various B-Raf and MEK inhibitors. Several of these trials are now in Phase III.
In this chapter, we will discuss new patents and patent applications related to inhibitors of the Ras/Raf/MEK/ERK pathway and some recent clinical trial results.
32) http://www.ncbi.nlm.nih.gov/
Biol Pharm Bull. 2011;34(12):1781-4.
Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Kohno M1, Tanimura S, Ozaki K.
1Laboratory of Cell Regulation, Department of Pharmaceutical Sciences, Graduate School of Biomedical Sciences, Nagasaki University, Japan
The extracellular signal-regulated kinase (ERK) pathway is a major determinant in the control of diverse cellular processes such as proliferation, survival, and motility. This pathway is often upregulated in human cancers and as such represents an attractive target for mechanism-based approaches to cancer treatment. However, specific blockade of the ERK pathway alone induces mostly cytostatic rather than proapoptotic effects, resulting in limited therapeutic efficacy. Blockade of the constitutively activated ERK pathway by an ERK kinase (MEK) inhibitor sensitizes tumor cells to apoptotic cell death induced by several cytotoxic anticancer agents including microtubule-destabilizing agents and histone deacetylase inhibitors, not only in vitro but also in tumor zenografts in vivo. Thus, low concentrations of these anticancer drugs that by themselves show little cytotoxicity effectively kill tumor cells in which the ERK pathway is constitutively activated when co-administrated with a MEK inhibitor. The combination of a cytostatic signaling pathway inhibitor (MEK inhibitors) and conventional anticancer drugs (microtubule-destabilizing agents or histone deacetylase inhibitors) provides an excellent basis for the development of safer anticancer chemotherapies with enhanced efficacy through lowering the required dose of the latter cytotoxic drugs.
33) http://www.ncbi.nlm.nih.gov/
Recent Pat Anticancer Drug Discov. 2009 Jan;4(1):28-35.
Recent developments in anti-cancer agents targeting the Ras/Raf/ MEK/ERK pathway. Wong KK.
The Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) pathway mediates cellular responses to different growth signals and is frequently deregulated in cancer. There are three Raf kinases-A-Raf, B-Raf, and C-Raf; however, only B-Raf is frequently mutated in various cancers. The most common B-Raf mutation involves a substitution of a glutamic acid residue to a valine moiety at codon 600. Subsequently, the MAPK pathway is constitutively activated, even in the absence of any growth signals. Although early attempts to target Ras have not yielded any viable drug candidates, many novel compounds inhibiting the activities of Raf and MEK have been developed and investigated in clinical trials in recent years. The first MEK inhibitor (CI-1040) lacked efficacy in clinical trials, but its low toxicity has encouraged the search for novel compounds with enhanced target potency to inhibit MAPK activation at low nanomolar concentrations. In this review, we will discuss new patents or patent applications related to inhibitors of the Ras/Raf/MEK/ERK pathway.
34) http://www.ncbi.nlm.nih.gov/
Ann Med. 2006;38(3):200-11.
Targeting the ERK signaling pathway in cancer therapy.
Kohno M1, Pouyssegur J.
Laboratory of Cell Regulation, Department of Pharmaceutical Sciences, Graduate School of Biomedical Sciences, Nagasaki University, Bunkyo-machi, Japan.
The extracellular signal-regulated kinase (ERK) signaling pathway is a major determinant in the control of diverse cellular processes such as proliferation, survival, differentiation and motility. This pathway is often up-regulated in human tumors and as such represents an attractive target for the development of anticancer drugs. Because of its multiple roles in the acquisition of a complex malignant phenotype, specific blockade of the ERK pathway is expected to result in not only an anti-proliferative effect but also in anti-metastatic and anti-angiogenic effects in tumor cells. Recently potent small-molecule inhibitors targeting the components of the ERK pathway have been developed. Among them, BAY 43-9006 (Raf inhibitor), and PD184352, PD0325901 and ARRY-142886 (MEK1/2 inhibitors) have reached the clinical trial stage. We briefly discuss the possibility that combination of ERK pathway inhibitors (cytostatic agents) and conventional anticancer drugs (cytotoxic agents) provides an excellent basis for the development of new chemotherapeutic strategies against cancer.
35) full free pdf
http://www.ncbi.nlm.nih.gov/
Life Sci. 2006 Jan 2;78(6):549-63. Epub 2005 Aug 18.
Cannabinoid signalling. Demuth DG1, Molleman A.
After their discovery, the two known cannabinoid receptors, CB(1) and CB(2), have been the focus of research into the cellular signalling mechanisms of cannabinoids. The initial assessment, mainly derived from expression studies, was that cannabinoids, via G(i/o) proteins, negatively modulate cyclic AMP levels, and activate inward rectifying K(+) channels. Recent findings have complicated this assessment on different levels: (1) cannabinoids include a wide range of compounds with varying profiles of affinity and efficacy at the known CB receptors, and these profiles do not necessarily match their biological activity; (2) CB receptors appear to be intrinsically active and possibly coupled to more than one type of G protein; (3) CB receptor signalling mechanisms are diverse and dependent on the system studied; (4) cannabinoids have other targets than CB receptors. The aim of this mini review is to discuss the current literature regarding CB receptor signalling pathways. These include regulation of adenylyl cyclase, MAP kinase, intracellular Ca(2+), and ion channels. In addition, actions of cannabinoids that are not mediated by CB(1) or CB(2) receptors are discussed.
——–
Manual Guzman
In the present work, we investigated how the CB1receptor is coupled to ERK activation and its implications in cannabinoid regulation of glial cell death/survival decision.
36) http://www.ncbi.nlm.nih.gov/
Mol Pharmacol. 2002 Dec;62(6):1385-92.
Mechanism of extracellular signal-regulated kinase activation by the CB(1) cannabinoid receptor.
Galve-Roperh I1, Rueda D, Gómez del Pulgar T, Velasco G, Guzmán M.
Cannabinoids, the active components of marijuana and their endogenous counterparts, exert many of their actions in brain through the seven-transmembrane receptor CB(1). This receptor is coupled to the activation of the extracellular signal-regulated kinase (ERK) cascade. However, the precise molecular mechanism for CB(1)-mediated ERK activation is still unknown. Here, we show that in U373 MG human astrocytoma cells, CB(1) receptor activation with the cannabinoid agonist delta(8)-tetrahydrocannabinol dimethyl heptyl (HU-210) was coupled to ERK activation and protection from ceramide-induced apoptosis. HU-210-induced ERK activation was inhibited by tyrphostin AG1478 and PP2, widely employed inhibitors of the epidermal growth factor receptor (EGF(R)) and the Src family of cytosolic tyrosine kinases, respectively. However, HU-210 stimulation resulted in neither EGF(R) phosphorylation, Src tyrosine phosphorylation, nor increased Src activity. In addition, dominant-negative forms of both proteins were unable to prevent cannabinoid-induced ERK activation, thus excluding the existence of CB(1)-mediated EGF(R) transactivation or Src activation. Wortmannin and 2-(4-morpholinyl)-8-phenyl-4H-
————-
Paola Massi
37) http://www.plosone.org/
Cannabidiol, a Non-Psychoactive Cannabinoid Compound, Inhibits Proliferation and Invasion in U87-MG and T98G Glioma Cells through a Multitarget Effect Marta Solinas, Paola Massi, Valentina Cinquina, Marta Valenti, Daniele Bolognini, Marzia Gariboldi, Elena Monti, Tiziana Rubino,
Daniela Parolaro mail
——-
38) http://www.ncbi.nlm.nih.gov/
Ann Med. 2006;38(3):200-11.
Targeting the ERK signaling pathway in cancer therapy.
Kohno M1, Pouyssegur J.
1Laboratory of Cell Regulation, Department of Pharmaceutical Sciences, Graduate School of Biomedical Sciences, Nagasaki University, Bunkyo-machi, Japan.
The extracellular signal-regulated kinase (ERK) signaling pathway is a major determinant in the control of diverse cellular processes such as proliferation, survival, differentiation and motility. This pathway is often up-regulated in human tumors and as such represents an attractive target for the development of anticancer drugs. Because of its multiple roles in the acquisition of a complex malignant phenotype, specific blockade of the ERK pathway is expected to result in not only an anti-proliferative effect but also in anti-metastatic and anti-angiogenic effects in tumor cells. Recently potent small-molecule inhibitors targeting the components of the ERK pathway have been developed. Among them, BAY 43-9006 (Raf inhibitor), and PD184352, PD0325901 and ARRY-142886 (MEK1/2 inhibitors) have reached the clinical trial stage. We briefly discuss the possibility that combination of ERK pathway inhibitors (cytostatic agents) and conventional anticancer drugs (cytotoxic agents) provides an excellent basis for the development of new chemotherapeutic strategies against cancer.
39) http://www.ncbi.nlm.nih.gov/
Biol Pharm Bull. 2011;34(12):1781-4.
Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Kohno M1, Tanimura S, Ozaki K.
1Laboratory of Cell Regulation, Department of Pharmaceutical Sciences, Graduate School of Biomedical Sciences, Nagasaki University, Japan.
The extracellular signal-regulated kinase (ERK) pathway is a major determinant in the control of diverse cellular processes such as proliferation, survival, and motility. This pathway is often upregulated in human cancers and as such represents an attractive target for mechanism-based approaches to cancer treatment. However, specific blockade of the ERK pathway alone induces mostly cytostatic rather than proapoptotic effects, resulting in limited therapeutic efficacy. Blockade of the constitutively activated ERK pathway by an ERK kinase (MEK) inhibitor sensitizes tumor cells to apoptotic cell death induced by several cytotoxic anticancer agents including microtubule-destabilizing agents and histone deacetylase inhibitors, not only in vitro but also in tumor zenografts in vivo. Thus, low concentrations of these anticancer drugs that by themselves show little cytotoxicity effectively kill tumor cells in which the ERK pathway is constitutively activated when co-administrated with a MEK inhibitor. The combination of a cytostatic signaling pathway inhibitor (MEK inhibitors) and conventional anticancer drugs (microtubule-destabilizing agents or histone deacetylase inhibitors) provides an excellent basis for the development of safer anticancer chemotherapies with enhanced efficacy through lowering the required dose of the latter cytotoxic drugs.
Paola Massi,
40) http://www.plosone.org/
Cannabidiol, a Non-Psychoactive Cannabinoid Compound, Inhibits Proliferation and Invasion in U87-MG and T98G Glioma Cells through a Multitarget Effect
Marta Solinas, Paola Massi, Valentina Cinquina, Marta Valenti, Daniele Bolognini, Marzia Gariboldi, Elena Monti, Tiziana Rubino, Daniela Parolaro mail
——-colon cancer utaly
Naples, Italy; Endocannabinoid Research Group, Italy.
41) http://www.ncbi.nlm.nih.gov/
Phytomedicine. 2014 Apr 15;21(5):631-9. doi: 10.1016/j.phymed.2013.11.006. Epub 2013 Dec 25.
Inhibition of colon carcinogenesis by a standardized Cannabis sativa extract with high content of cannabidiol. Romano B1, Borrelli F2, Pagano E2, Cascio MG3, Pertwee RG3, Izzo AA4.
Author information 1Department of Pharmacy, University of Naples Federico II, Naples, Italy; Endocannabinoid Research Group, Italy; School of Medical Sciences, Institute of Medical Sciences, University of Aberdeen, Aberdeen AB25 2ZD, United Kingdom.
2Department of Pharmacy, University of Naples Federico II, Naples, Italy; Endocannabinoid Research Group, Italy.
3School of Medical Sciences, Institute of Medical Sciences, University of Aberdeen, Aberdeen AB25 2ZD, United Kingdom.
4Department of Pharmacy, University of Naples Federico II, Naples, Italy; Endocannabinoid Research Group, Italy.
Colon cancer is a major public health problem. Cannabis-based medicines are useful adjunctive treatments in cancer patients. Here, we have investigated the effect of a standardized Cannabis sativa extract with high content of cannabidiol (CBD), here named CBD BDS, i.e. CBD botanical drug substance, on colorectal cancer cell proliferation and in experimental models of colon cancer in vivo.
METHODS:
Proliferation was evaluated in colorectal carcinoma (DLD-1 and HCT116) as well as in healthy colonic cells using the MTT assay. CBD BDS binding was evaluated by its ability to displace [(3)H]CP55940 from human cannabinoid CB1 and CB2 receptors. In vivo, the effect of CBD BDS was examined on the preneoplastic lesions (aberrant crypt foci), polyps and tumours induced by the carcinogenic agent azoxymethane (AOM) as well as in a xenograft model of colon cancer in mice.
RESULTS:
CBD BDS and CBD reduced cell proliferation in tumoral, but not in healthy, cells. The effect of CBD BDS was counteracted by selective CB1 and CB2 receptor antagonists. Pure CBD reduced cell proliferation in a CB1-sensitive antagonist manner only. In binding assays, CBD BDS showed greater affinity than pure CBD for both CB1 and CB2 receptors, with pure CBD having very little affinity. In vivo, CBD BDS reduced AOM-induced preneoplastic lesions and polyps as well as tumour growth in the xenograft model of colon cancer.
CONCLUSIONS:
CBD BDS attenuates colon carcinogenesis and inhibits colorectal cancer cell proliferation via CB1 and CB2 receptor activation. The results may have some clinical relevance for the use of Cannabis-based medicines in cancer patients.
==============================
Manual Guzman Ceramide Spain
Manual guzman Pancreatic CA
42) http://cancerres.aacrjournals.org/content/66/13/6748.full.html
Cannabinoids Induce Apoptosis of Pancreatic Tumor Cells via Endoplasmic Reticulum Stress–Related Genes Cancer Res. 2006 Jul 1;66(13):6748-55.
Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Carracedo A1, Gironella M, Lorente M, Garcia S, Guzmán M, Velasco G, Iovanna JL. 1Department of Biochemistry and Molecular Biology I, School of Biology, Complutense University, c/José Antonio Novais s/n, 28040 Madrid, Spain.
Pancreatic adenocarcinomas are among the most malignant forms of cancer and, therefore, it is of especial interest to set new strategies aimed at improving the prognostic of this deadly disease. The present study was undertaken to investigate the action of cannabinoids, a new family of potential antitumoral agents, in pancreatic cancer. We show that cannabinoid receptors are expressed in human pancreatic tumor cell lines and tumor biopsies at much higher levels than in normal pancreatic tissue. Studies conducted with MiaPaCa2 and Panc1 cell lines showed that cannabinoid administration (a) induced apoptosis, (b) increased ceramide levels, and (c) up-regulated mRNA levels of the stress protein p8. These effects were prevented by blockade of the CB(2) cannabinoid receptor or by pharmacologic inhibition of ceramide synthesis de novo. Knockdown experiments using selective small interfering RNAs showed the involvement of p8 via its downstream endoplasmic reticulum stress-related targets activating transcription factor 4 (ATF-4) and TRB3 in Delta(9)-tetrahydrocannabinol-
Pancreatic cancer is one of the most malignant and aggressive forms of cancer ( 1). With an incidence of 10/10,000 for men and 7/10,000 for women, it represents the fourth most common death-causing cancer in the United States ( 2) and the fifth in the Western world overall ( 3). About 95% of pancreatic cancers cases are ductal adenocarcinomas. The anatomic localization of the pancreas and the nonspecific nature of the symptoms result in a complex and delayed diagnosis. Therefore, at the time of detection, 85% of patients show metastasic infiltrations in proximal lymphatic nodes, liver, or lungs, and only 15% to 20% of the tumors are typically found resectable ( 1). In addition, <20% of the operated patients survive up to 5 years. Treatment of unresectable tumors is currently based on administration of fluorouracil chemoradiation for locally advanced tumors and gemcitabine chemotherapy for metastatic disease ( 1). However, despite maximal optimization of these therapies, the median survival for the affected patients remains ∼1 year. This antitumoral action of cannabinoids relies, at least in part, on the ability of these compounds to directly affect the viability, via induction of apoptosis or cell cycle arrest, of a wide spectrum of tumor cells in culture
THC induces apoptosis of pancreatic tumor cells.
In the present report, we show that cannabinoids induce apoptosis of pancreatic tumor cell lines in vitro and exert a remarkable growth-inhibiting effect in models of pancreatic cancer in vivo.
Of potential interest for future cannabinoid-based therapies, cannabinoid treatment does not seem to activate this pathway in normal pancreas or spleen, suggesting that these agents may activate the endoplasmic reticulum stress proapoptotic pathway selectively in tumor cells.
Thus, our findings indicate that the mechanism of cannabinoid-induced apoptosis of human pancreatic tumor cells involves a CB2 receptor–dependent accumulation of de novo synthesized ceramide that leads to p8, ATF-4, and TRB3 up-regulation
43) http://www.ncbi.nlm.nih.gov/
Life Sci. 2005 Aug 19;77(14):1723-31.
Cannabinoids and ceramide: two lipids acting hand-by-hand.
Velasco G1, Galve-Roperh I, Sánchez C, Blázquez C, Haro A, Guzmán M.
1Department of Biochemistry and Molecular Biology I, School of Biology, Complutense University, 28040 Madrid, Spain.
Cannabinoids, the active components of Cannabis sativa (marijuana) and their endogenous counterparts, exert their effects by binding to specific G-protein-coupled receptors that modulate adenylyl cyclase and ion channels. Recent research has shown that the CB1 cannabinoid receptor is also coupled to the generation of the lipid second messenger ceramide via two different pathways: sphingomyelin hydrolysis and ceramide synthesis de novo. Sustained ceramide accumulation in tumor cells mediates cannabinoid-induced apoptosis, as evidenced by in vitro and in vivo studies. This effect seems to be due to the impact of ceramide on key cell signalling systems such as the extracellular signal-regulated kinase cascade and the Akt pathway. These findings provide a new conceptual view on how cannabinoids act, and raise interesting physiological and therapeutic questions.
———————————————-
P8 Stress-inducible Protein p8
44) http://www.ncbi.nlm.nih.gov/
J Biol Chem. Jan 15, 2010; 285(3): 1577–1581.
Stress-inducible Protein p8 Is Involved in Several Physiological and Pathological Processes*Sandro Goruppi‡,1 and Juan Lucio Iovanna§,2
Perhaps the most important antitumor role for p8 is in mediating the cannabinoid-induced apoptosis of several kind of tumor cells, in particular glioblastomas (12). In glioblastoma cells, p8 is required for the efficient induction of the endoplasmic reticulum stress-related genes ATF4, CHOP, and TRB3 (Table 1) (12). This pathway is selectively activated only in transformed cells and not in non-transformed cells. p8 is also necessary for cannabinoid-induced autophagy and apoptosis (36).
===============
Manuel Guzmán Glioma 2009 THC autophagy double membrane
45) http://www.ncbi.nlm.nih.gov/pubmed/19425170
J Clin Invest. May 1, 2009; 119(5): 1359–1372.
Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells
María Salazar,1,2 Arkaitz Carracedo,1 Íñigo J. Salanueva,1 Sonia Hernández-Tiedra,1 Mar Lorente,1,2 Ainara Egia,1 Patricia Vázquez,3 Cristina Blázquez,1,2 Sofía Torres,1 Stephane García,4 Jonathan Nowak,4 Gian María Fimia,5 Mauro Piacentini,5 Francesco Cecconi,6 Pier Paolo Pandolfi,7 Luis González-Feria,8 Juan L. Iovanna,4 Manuel Guzmán,1,2 Patricia Boya,3 and Guillermo Velasco1,2
Autophagy can promote cell survival or cell death, but the molecular basis underlying its dual role in cancer remains obscure. Here we demonstrate that Δ9-tetrahydrocannabinol (THC), the main active component of marijuana, induces human glioma cell death through stimulation of autophagy. Our data indicate that THC induced ceramide accumulation and eukaryotic translation initiation factor 2α (eIF2α) phosphorylation and thereby activated an ER stress response that promoted autophagy via tribbles homolog 3–dependent (TRB3-dependent) inhibition of the Akt/mammalian target of rapamycin complex 1 (mTORC1) axis. We also showed that autophagy is upstream of apoptosis in cannabinoid-induced human and mouse cancer cell death and that activation of this pathway was necessary for the antitumor action of cannabinoids in vivo. These findings describe a mechanism by which THC can promote the autophagic death of human and mouse cancer cells and provide evidence that cannabinoid administration may be an effective therapeutic strategy for targeting human cancers.
Macro-autophagy, hereafter referred to as “autophagy,” is a highly conserved cellular process in which cytoplasmic materials — including organelles — are sequestered into double-membrane vesicles called autophagosomes and delivered to lysosomes for degradation or recycling
we performed electron microscopy analysis of U87MG human astrocytoma cells. Interestingly, double membrane vacuolar structures with the morphological features of autophagosomes were observed in THC-treated cells.
—————————————————————
cannabinoids for Pain
46) http://www.ncbi.nlm.nih.gov/
Ther Clin Risk Manag. 2008 Feb;4(1):245-59.
Cannabinoids in the management of difficult to treat pain.
Russo EB. GW Pharmaceuticals Vashon, WA, USA.
This article reviews recent research on cannabinoid analgesia via the endocannabinoid system and non-receptor mechanisms, as well as randomized clinical trials employing cannabinoids in pain treatment. Tetrahydrocannabinol (THC, Marinol((R))) and nabilone (Cesamet((R))) are currently approved in the United States and other countries, but not for pain indications. Other synthetic cannabinoids, such as ajulemic acid, are in development. Crude herbal cannabis remains illegal in most jurisdictions but is also under investigation. Sativex((R)), a cannabis derived oromucosal spray containing equal proportions of THC (partial CB(1) receptor agonist ) and cannabidiol (CBD, a non-euphoriant, anti-inflammatory analgesic with CB(1) receptor antagonist and endocannabinoid modulating effects) was approved in Canada in 2005 for treatment of central neuropathic pain in multiple sclerosis, and in 2007 for intractable cancer pain. Numerous randomized clinical trials have demonstrated safety and efficacy for Sativex in central and peripheral neuropathic pain, rheumatoid arthritis and cancer pain. An Investigational New Drug application to conduct advanced clinical trials for cancer pain was approved by the US FDA in January 2006. Cannabinoid analgesics have generally been well tolerated in clinical trials with acceptable adverse event profiles. Their adjunctive addition to the pharmacological armamentarium for treatment of pain shows great promise.
Brain plasticity- progenitor cells in hippocampus
47) Hallmarks_of_Cancer_Cell_Jan_2000
Cell. 2000 Jan 7;100(1):57-70. The hallmarks of cancer.Hanahan D1, Weinberg RA. Department of Biochemistry, Hormone Research Institute, University of California at San Francisco, 94143, USA.
——————
Malignant Brain Glioma Cells
U87-MG cell line derives from glioblastoma/astrocytoma, whereas T98G from glioblastoma multiforme.
Besides the wide inhibition of different proteins, CBD strongly down-regulated two signaling pathways critical for glioma cell survival and proliferation, such as ERK and PI3K/Akt. These signaling molecules have been shown to play a crucial role in tumor cell proliferation and in cell escape from apoptosis or cycle arrest as well as in cell motility and invasion [37], [38]. Similar results were recently obtained by Soroceanu et al. (2013) in U251 cells, demonstrating a CBD-dependent ERK and Akt downregulation in glioma cells, together with a decrease in MMP-2 level and in invasiveness. They suggest that CBD may act through modulation of Id-1, a transcription factor regulator that seems to be involved in glioma malignancy [39].
—————————————
http://www.greenmedinfo.com/
references
Crosstalk between chemokine receptor CXCR4 and cannabinoid receptor CB2 in modulating breast cancer growth and invasion. Nasser MW; et al; PLoS One. 2011;6(9):e23901. doi: 10.1371/journal.pone.0023901. Epub 2011 Sep 7; http://www.ncbi.nlm.nih.gov/
Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells; McAllister SD et al; Mol Cancer Ther. 2007 Nov;6(11):2921-7;
Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation; Caffarel MM et al; Cancer Res; 2006 Jul 1;66(13):6615-21;
Cannabinoids: a new hope for breast cancer therapy? Caffarel MM et al; Cancer Treat Rev.: 2012 Nov; 38(7):911-8. doi: 10.1016/j.ctrv.2012.06.005. Epub 2012 Jul 7;
Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. Ramer R et al; FASEB J.; 2012 Apr;26(4):1535-48. doi: 10.1096/fj.11-198184. Epub 2011 Dec 23;
Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis; Preet A, et al; Cancer Prev Res (Phila). 2011 Jan; 4(1):65-75. doi: 10.1158/1940-6207.CAPR-10-
Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo; A Preetet al; Oncogene; (2008) 27,339–346; doi:10.1038/sj.onc.1210641; published online 9 July 2007;
The role of cannabinoids in prostate cancer: Basic science perspective and potential clinical applications; Juan A. Ramos and Fernando J. Bianco; Indian J Urol. 2012 Jan-Mar; 28(1): 9–14;.doi:10.4103/0970-1591.
Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. Aviello G et al; ;J Mol Med (Berl); 2012 Aug;90(8):925-34. doi: 10.1007/s00109-011-0856-x. Epub 2012; Jan 10.;
Cannabinoid receptors as a target for therapy of ovarian cancer. Farrukh Afaq; et al;, Proc Amer Assoc Cancer Res, Volume 47, 2006;
Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. McKallip RJ et al; Blood. 2002 Jul 15;100(2):627-34.;
Delta9-tetrahydrocannabinol-
Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. Casanova ML et al: J Clin Invest. 2003 Jan;111(1):43-50;
Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Vara D et al; Cell Death Differ; 2011 Jul;18(7):1099-111. doi: 10.1038/cdd.2011.32. Epub 2011 Apr 8.;
The dual effects of delta(9)-tetrahydrocannabinol on cholangiocarcinoma cells: anti-invasion activity at low concentration and apoptosis induction at high concentration. Leelawat Set al;Cancer Invest. 2010 May;28(4):357-63. doi: 10.3109/07357900903405934;
Cannabis-induced cytotoxicity in leukemic cell lines: the role of the cannabinoid receptors and the MAPK pathway; Powles T et al; Blood;.2005 Feb 1;105(3):1214-21; Epub 2004 Sep 28.;
In vivo effects of cannabinoids on macromolecular biosynthesis in Lewis lung carcinomas; Friedman MA; Cancer Biochem Biophys. 1977;2(2):51-4.;
Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases-1; Ramer Ret al; Biochem Pharmacol; 2010 Apr 1;79(7):955-66. doi: 10.1016/j.bcp.2009.11.007. Epub 2009 Nov 13;
Cannabinoids in intestinal inflammation and cancer. Izzo AA1, Camilleri M.; Pharmacol Res; 2009 Aug;60(2):117-25. doi: 10.1016/j.phrs.2009.03.008. Epub 2009 Mar 18;
Involvement of cannabinoids in cellular proliferation; López-Rodríguez ML et al; Mini Rev Med Chem; 2005 Jan;5(1):97-106
Jeffrey Dach MD
7450 Griffin Road, Suite 190
Davie, Fl 33314
954-792-4663
http://www.jeffreydachmd.com
http://www.drdach.com
http://www.naturalmedicine101.com
Click Here for: Dr Dach’s Online Store for Pure Encapsulations Supplements
Click Here for: Dr Dach’s Online Store for Nature’s Sunshine Supplements
Web Site and Discussion Board Links:
http://jdach1.typepad.com/blog/
http://disc.yourwebapps.com/Indices/244124.html
http://disc.yourwebapps.com/Indices/244066.html
http://disc.yourwebapps.com/Indices/244067.html
http://disc.yourwebapps.com/Indices/244161.html
http://disc.yourwebapps.com/Indices/244163.html
Disclaimer click here: http://www.drdach.com/wst_page20.html
The reader is advised to discuss the comments on these pages with his/her personal physicians and to only act upon the advice of his/her personal physician. Also note that concerning an answer which appears as an electronically posted question, I am NOT creating a physician — patient relationship. Although identities will remain confidential as much as possible, as I can not control the media, I can not take responsibility for any breaches of confidentiality that may occur.
Copyright (c) 2014 Jeffrey Dach MD All Rights Reserved. This article may be reproduced on the internet without permission, provided there is a link to this page and proper credit is given.
FAIR USE NOTICE: This site contains copyrighted material the use of which has not always been specifically authorized by the copyright owner. We are making such material available in our efforts to advance understanding of issues of significance. We believe this constitutes a ‘fair use’ of any such copyrighted material as provided for in section 107 of the US Copyright Law. In accordance with Title 17 U.S.C. Section 107, the material on this site is distributed without profit to those who have expressed a prior interest in receiving the included information for research and educational purposes.



How Does Cannabis Kill Cancer Cells Part Two - Jeffrey Dach MD October 24, 2014 at 1:59 PM
[…] ← Previous […]
20 orvosbiológiai tanulmány bizonyítja a kannabisz rákölő tulajdonságait - Medical Marijuana - Kender Fórum December 16, 2014 at 4:29 PM
[…] How Does Cannabis Kill Cancer Cells ? Válaszol idézettel […]